Executive Summary
On April 18, 2026, President Trump signed the Accelerating Medical Treatments for Serious Mental Illness Executive Order, directing federal agencies to fast-track development and patient access for psychedelic medicines. The order is best understood as a federal acceleration directive: it does not legalize or reschedule any substance, nor does it create new appropriations, but it establishes five concrete mechanisms that could meaningfully reshape the field.
What the Order Does
The order’s five operative provisions are:
- FDA Priority Vouchers: Priority access to the FDA’s expedited Commissioner’s National Priority Voucher program for psychedelic drugs with Breakthrough Therapy designation, compressing review timelines from roughly 12 months to 1-2 months.
- Right to Try Pathway: A directive to the FDA and DEA to create a Right to Try pathway for eligible patients to access investigational psychedelics, including ibogaine, by establishing new Schedule I handling authorizations for physicians.
- Federal-State Matching Funds: A $50 million federal-state matching fund through ARPA-H for state-level psychedelic research programs, with Texas’s $50 million ibogaine research consortium as the explicit model.
- Inter-Agency Data Sharing: Formal data-sharing mandates between HHS, FDA, the VA, and the private sector to feed clinical evidence into the FDA’s evaluation pipeline.
- Timely Rescheduling Pathway: A directive to the Attorney General to fast-track DEA rescheduling reviews for any Schedule I psychedelic that successfully completes Phase 3 trials and receives FDA approval.
What the Order Does Not Do
Critically, the order does not reschedule ibogaine, psilocybin, MDMA, or LSD; all remain Schedule I. It draws on existing ARPA-H funds rather than creating new appropriations. It contains a standard non-enforcement clause (Sec. 6(c)), meaning no patient or developer can sue to compel agency action under it. It does not:
- Change FDA’s evidence standards for approval
- Address the domestic supply chain for ibogaine
- Address coverage, reimbursement, or clinical delivery infrastructure, which are the gaps that will ultimately determine whether approved treatments reach patients at scale

The Underlying Context
The order responds to documented unmet need:
- Veterans: Over 6,000 veteran suicides annually, a rate more than double the civilian average
- Mental illness: More than 14 million American adults with serious mental illness for whom standard treatments are often insufficient
- Opioid crisis: Despite a significant decline in overdose deaths in 2024, the crisis still claims roughly 54,000 lives per year
Published clinical research, particularly Stanford’s 2024 Nature Medicine ibogaine study in veterans with traumatic brain injury, has provided evidence supporting the urgency.
The State Landscape
The federal order lands into substantial pre-existing state-level momentum:
- Texas has committed $50 million to an ibogaine research consortium led by UTHealth Houston and UT Galveston
- Mississippi, Kentucky, Utah, and Arizona have passed or advanced similar legislation
- Trigger laws, which would automatically reschedule psychedelics at the state level upon federal action, are advancing in multiple states
The federal $50 million matching structure appears designed in part to close the private-sector funding gap Texas encountered when no drug company met the state’s matching requirements.
The Pipeline
The rescheduling pathway in Section 5 is most immediately relevant to psilocybin and LSD-derived therapies:
- Compass Pathways announced successful Phase 3 results for COMP360 psilocybin in treatment-resistant depression in February 2026 (Compass has now also received FDA rolling NDA review and a Commissioner’s National Priority Voucher for COMP360, making it the clearest near-term test case for the Order’s FDA acceleration mechanism.)
- Definium (MindMed) has three Phase 3 trials underway for its LSD-derived MM120
- Ibogaine candidates remain several years from Phase 3 completion, though FDA’s concurrent IND clearance for ibogaine opens US-based trials for the first time
What Remains Unresolved
Key implementation risks include:
- DEA follow-through on Schedule I access pathways (the agency has already missed statutory deadlines under the 2025 HALT Fentanyl Act)
- Funding durability: the ARPA-H allocation is a reallocation from existing funds, not a new appropriation, and is subject to future budget pressure
- Ibogaine supply chain: no domestic supply of pharmaceutical-grade ibogaine exists at scale; Nagoya Protocol-compliant sourcing from Gabon remains limited
- Cardiac safety: ibogaine’s documented risk of QT prolongation and incomplete US Phase 1 evidence base constrain the Right to Try pathway in practice
- Coverage and delivery gaps: no framework exists yet for reimbursement, REMS design, clinical training, or delivery infrastructure; these decisions will determine whether approved therapies are broadly accessible or confined to specialized, out-of-pocket settings
- Political durability: a subsequent administration could rescind the order; Congressional action remains necessary for structural change
The Ibogaine Healthcare Policy Institute (IHPI)
The Ibogaine Healthcare Policy Institute is a neutral, evidence-based, implementation-focused nonprofit institute working at the intersection of psychedelic medicine, healthcare policy, and public health. IHPI’s mission is to move ibogaine from promise to policy to practice, with a specific focus on the coverage and delivery infrastructure that will help ensure that clinical evidence translates into responsible patient access.
IHPI’s work is organized around four areas that directly address the unresolved gaps identified above:
- Coverage playbooks: Developing patient eligibility criteria, prior authorization rules, and medical coding pathways that payers and Medicaid programs need to make early coverage decisions
- Health economics: Building cost-offset analyses, utilization scenarios, and risk frameworks for payer integration into benefits, budgeting, and procurement workflows
- Delivery blueprints: Mapping residential, outpatient, and hospital pathways for safe delivery, including staffing models, screening protocols, cardiac monitoring requirements, and quality assurance processes
- Veterans-first pilots: Structured pilots with the veteran cohort, in coordination with VA-adjacent partners and community providers, to generate the operational evidence base that broader implementation requires
IHPI’s board brings cross-sector experience spanning clinical addiction medicine (Andrea G. Barthwell, MD, DFASAM, former Deputy Director at the White House Office of National Drug Control Policy), federal health agency operations (Leith States, MD, MPH, MBA, former Acting Assistant Secretary for Health at HHS), and healthcare public-private partnerships (Eric Letsinger, Founder of Quantified Ventures).
IHPI was founded in February of 2026 and is actively engaging with payers, health systems, veterans organizations, federal partners, and therapeutic developers. More information is available at delphi-circle.com/ihpi.
Bottom Line
The Executive Order is a meaningful shift in federal posture and the most significant federal action on psychedelic medicine to date. Its impact will be determined by downstream decisions made by agencies, payers, Congress, and clinical delivery systems that the order neither directs nor funds. The gap between regulatory acceleration and actual patient access remains wide, and closing it requires implementation work that has not yet begun in earnest. For a more detailed and nuanced analysis, continue reading below.
Index
- Executive Summary
- Section 1: Introduction
- Section 2: What the EO Actually Does
- Section 3: What the EO Does Not Do
- Section 4: Why This Matters — The Underlying Need
- Section 5: The State-Level Story
- Section 6: What Access Actually Looks Like for Patients
- Section 7: What Remains Unresolved
- Section 8: Where the Ibogaine Healthcare Policy Institute (IHPI) Fits
- Section 9: Sources and Further Reading
Section 1: Introduction
More than 14 million American adults live with serious mental illness. Roughly 8 million of them rely on prescription medication for these conditions. For many patients, those medications work. For a substantial share, they do not.
The numbers among veterans are starker. The United States has lost more than 6,000 veterans to suicide every year for over two decades. The veteran suicide rate runs more than twice the rate among non-veteran adults.
For patients whose conditions persist after standard treatment, the field has produced little meaningful innovation in a generation.
On Saturday, April 18, 2026, President Trump signed an Executive Order titled Accelerating Medical Treatments for Serious Mental Illness. It directs federal agencies to fast-track FDA review of psychedelic drugs with established clinical promise, expand patient access under the Right to Try Act, fund state-level research programs, and prepare a pathway for rescheduling. The order names ibogaine compounds explicitly.
The order is best understood as a federal acceleration directive. It does not legalize or reschedule any psychedelic substance, and it brings no new federal appropriations. The mechanisms it establishes are concrete and operational. If implemented, they will change the landscape for patients, researchers, and developers in the years ahead.
What follows is a clear-eyed read of what the order does, what it does not do, what is most likely to matter in practice, and what remains unresolved.
Section 2: What the EO Actually Does
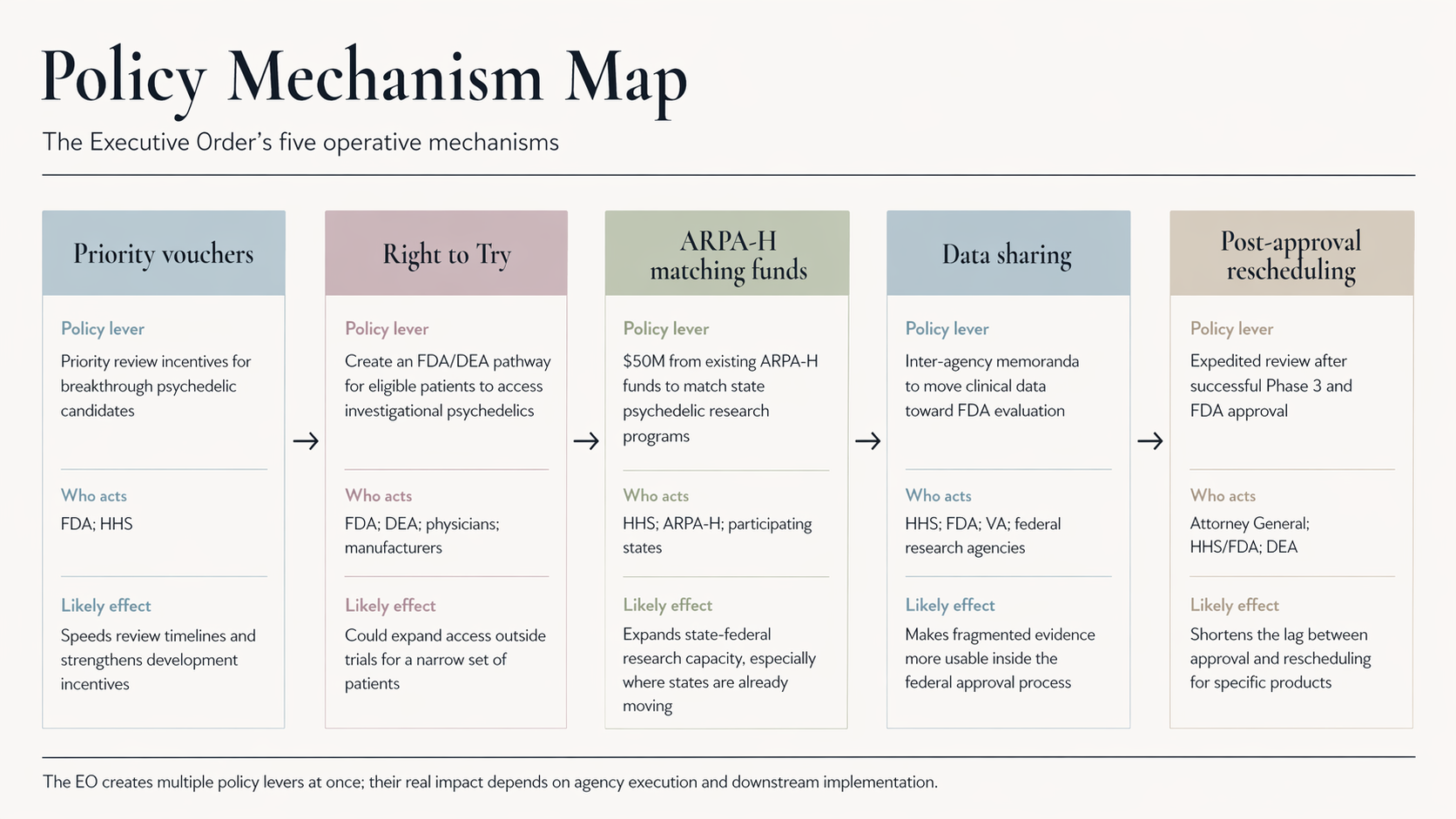
The Executive Order contains five operative mechanisms across four substantive sections. None of them legalizes any psychedelic substance. Each removes a specific regulatory, funding, or coordination barrier that has slowed psychedelic medicine development and patient access in recent years.
The five mechanisms are:
- FDA Priority Voucher access for psychedelic drugs holding Breakthrough Therapy designation (Section 2.1)
- A DEA-cleared Right to Try pathway for eligible patients (Sec. 2(b))
- $50 million in federal-state matching funds through ARPA-H for state-level programs (Sec. 3)
- A mandate for HHS, FDA, the VA, and the private sector to share data and increase clinical trial participation (Sec. 4)
- A timely-rescheduling pathway tied to successful Phase 3 completion (Sec. 5)
Each is addressed in turn below.
2.1 FDA Priority Vouchers
The first mechanism reaches into a program that did not exist a year ago.
In June 2025, FDA Commissioner Marty Makary launched the Commissioner’s National Priority Voucher pilot program. The program compresses the standard FDA new drug application review window from 10 to 12 months down to 1 to 2 months. It does this by replacing the standard sequential office-by-office review with a single multidisciplinary “tumor board style” meeting in which experts from across the agency review the application together over the course of a day.
By April 2026, the FDA had awarded 18 priority vouchers and reached 6 final decisions under the program. Awardees are selected based on alignment with national health priorities, including addressing a public health crisis, advancing innovative therapies, meeting large unmet medical needs, supporting domestic manufacturing, and improving affordability. The program remains in pilot status, with a public hearing on its design scheduled for June 4, 2026.
On April 24, 2026, six days after the Executive Order, FDA issued National Priority Vouchers to three companies studying psychedelic or psychedelic-adjacent treatments: psilocybin for treatment-resistant depression, psilocybin for major depressive disorder, and methylone for PTSD. FDA’s release named the indications rather than the sponsors. Compass Pathways confirmed that COMP360 received the voucher and that FDA granted rolling NDA review; Usona Institute confirmed that its psilocybin program received a voucher; and Transcend Therapeutics later confirmed that TSND-201 received a voucher for PTSD.
The April 18 Executive Order directs the Commissioner to provide priority vouchers “to appropriate psychedelic drugs that have received a Breakthrough Therapy designation and are in accordance with the criteria of the National Priority Voucher Program.”
This is significant because it stacks two acceleration mechanisms on top of each other. Breakthrough Therapy designation, established by Congress in 2012, already gives qualifying drugs access to intensive FDA guidance and rolling review during clinical development. Several psychedelic candidates already hold this status:
- MDMA, granted Breakthrough Therapy designation in 2017 for the treatment of post-traumatic stress disorder, originally to MAPS Public Benefit Corporation (now Lykos Therapeutics).
- Psilocybin, granted Breakthrough Therapy designation in two separate programs: Compass Pathways for treatment-resistant depression (2018) and the Usona Institute for major depressive disorder (2019).
- MM120, MindMed’s lysergide d-tartrate (an LSD formulation), was granted Breakthrough Therapy designation in March 2024 for generalized anxiety disorder.
- CYB003, Cybin’s deuterated psilocybin analog, was granted Breakthrough Therapy designation in 2024 as an adjunctive treatment for major depressive disorder.
- TSND-201, Transcend Therapeutics’ methylone program for PTSD, received Breakthrough Therapy designation in July 2025 and is now enrolling patients in a U.S. Phase 3 trial. Transcend describes TSND-201 as non-hallucinogenic and not active at 5-HT2A receptors, so it is best treated as a psychedelic-adjacent or empathogen/neuroplastogen program rather than a classic psychedelic.
- Esketamine (Spravato), Janssen’s intranasal NMDA receptor antagonist, granted Breakthrough Therapy designation twice: for treatment-resistant depression (November 2013) and for major depressive disorder with imminent risk for suicide (August 2016).
The Order does not change the scientific or regulatory standards by which the FDA reviews these candidates. It changes the queue. For developers who have spent years and hundreds of millions of dollars on Phase 3 work, that compression is meaningful. That point is now testable rather than theoretical. The CNPV program is explicitly nontransferable, offers enhanced communication and rolling review, and targets a 1–2-month review window rather than 6+ months, while maintaining the same statutory approval standards. Depending on the program’s stage, a voucher can now help align FDA interactions before submission and compress final review after filing. For Compass, this is already tied to rolling NDA review; for Usona and Transcend, it is better described as an acceleration signal for ongoing late-stage development rather than an imminent approval decision.
One caveat is worth surfacing: priority vouchers are awarded at the Commissioner’s discretion. The Order directs the Commissioner to award them to qualifying psychedelic candidates, but the determination of which candidates qualify, and when, remains an FDA judgment.
2.2 The Right to Try pathway for psychedelics
The second provision is likely to matter most for individual patients over the next two years. It is also the provision most likely to face implementation friction.
Section 2(b) of the Order directs the FDA and the DEA to “facilitate and establish a pathway for eligible patients to access psychedelic drugs, including ibogaine compounds, under the Right to Try Act (21 U.S.C. 360bbb-0a), including any necessary Schedule I handling authorizations for treating physicians and researchers, consistent with 21 U.S.C. 823, and any applicable waiver authority under the Controlled Substances Act.”
To understand why this matters, it helps to know what the Right to Try Act has and has not been able to do.
The federal Right to Try Act, signed by President Trump on May 30, 2018, established a pathway for patients with life-threatening conditions to access investigational drugs that have completed a Phase 1 clinical trial and remain in active development. The pathway sits outside the FDA’s existing Expanded Access program and was designed to let patients work directly with manufacturers, with the FDA’s involvement minimized.
In practice, the federal Act has produced limited results. By June 2019, one year after the law took effect, only two patients had reportedly accessed investigational treatments through the federal pathway.
For patients seeking access to Schedule I substances, including psychedelics, Right to Try has been functionally closed. The DEA has maintained that because Schedule I status by definition means a substance has “no currently accepted medical use,” no DEA registration exists that would authorize a treating physician to administer such substances under the Right to Try framework. In February 2025, the Ninth Circuit Court of Appeals affirmed this position in Advanced Integrated Medical Science Institute, PLLC v. DEA, ruling that the Right to Try Act did not impair the DEA’s regulatory authority over controlled substances. The case had been brought by a Washington State physician, Dr. Sunil Aggarwal, who had spent years seeking to use psilocybin in end-of-life cancer care.
Congress has attempted to fix this legislatively. The Right to Try Clarification Act, introduced in the House in 2023, did not advance. In late 2025, Senators Cory Booker and Rand Paul, together with Representatives Madeleine Dean and Nancy Mace, introduced the Freedom to Heal Act, which would amend the Controlled Substances Act to require the DEA to register and authorize qualified physicians to administer Schedule I drugs to eligible patients with life-threatening conditions. The bill remains pending.
The Executive Order is now directing the DEA to do administratively what Congress has not been able to do legislatively. This is the most consequential operational provision in the Order for individual patient access. If the DEA establishes a registration pathway that allows treating physicians to administer ibogaine, psilocybin, or other psychedelic compounds to eligible patients with life-threatening conditions, the Right to Try Act becomes a meaningfully different instrument than it has been for the last seven years.
Two cautions follow.
- First, the Order directs the DEA to establish the pathway. It does not specify how restrictive the registration process can be, which physicians and patients qualify, or how the supply will be assured for a population beyond the existing trial-enrolled cohort. The shape of the final pathway sits with the DEA.
- Second, this provision falls squarely within Section 6(c) of the Order, which states that the Order “is not intended to, and does not, create any right or benefit, substantive or procedural, enforceable at law or in equity by any party against the United States.” If the DEA chooses to slow-walk implementation, no patient or physician has standing to sue under the Order itself.
The mechanism is real. The follow-through is the question.
2.3 $50 million in federal-state matching funds
Of the five operative mechanisms in the Order, Section 3 has the clearest near-term operational lever.
Section 3 reads: “The Secretary of Health and Human Services shall, through the Advanced Research Projects Agency for Health, allocate at least $50 million from existing funds to support and partner with State governments that have enacted or are developing programs to advance psychedelic drugs for serious mental illnesses, including through Federal funding, technical assistance, and data sharing as appropriate and consistent with applicable law.”
Three elements in that paragraph deserve attention: ARPA-H as the funding vehicle, the federal-state matching structure, and the explicit naming of Texas in the accompanying White House Fact Sheet.
ARPA-H as the funding vehicle
The Advanced Research Projects Agency for Health was established in March 2022 as an agency within the Department of Health and Human Services. It operates outside the traditional NIH peer-review model and is explicitly modeled on DARPA: high-risk, high-impact research, run by a relatively small number of program managers with significant autonomy over their funding portfolios. The agency awards projects through Innovation Solutions Openings and Broad Agency Announcements rather than through the standard NIH grant cycle.
This matters for two reasons. First, ARPA-H exists specifically to take on problems that traditional research and commercial structures have failed to solve. Psychedelic medicine fits that frame. Second, ARPA-H’s funding mechanisms allow for faster, less constrained awards than traditional NIH grants, which is critical if the federal money is going to move on a timeline that matches state-level activity.
The federal-state matching structure
The funding is described as supporting “State governments that have enacted or are developing programs.” The structure puts the federal government in a partnership posture with states. It rewards jurisdictions that have already done the political work of passing their own legislation, by adding federal resources to existing state infrastructure.
That is also the structure most likely to generate concrete near-term clinical activity, because it builds on existing state-level momentum without waiting for a new federal program to be designed from scratch.
Why Texas is the named precedent
The accompanying White House Fact Sheet calls out one state by name: “In 2025, the state of Texas recognized this potential and launched a research consortium to accelerate ibogaine clinical trials and drug development.”
A short timeline of what Texas has done is worth walking through, because the federal Order can only be properly understood against it.
In May 2025, the Texas Legislature passed House Bill 3717 (authored by Rep. Cody Harris, R-Palestine) and Senate Bill 2308 (sponsored by Sen. Tan Parker, R-Flower Mound), authorizing $50 million in state funding for ibogaine clinical trials. Governor Greg Abbott signed SB 2308 into law on June 11, 2025. At the time, the state was widely described as having launched the largest publicly funded psychedelic research initiative in the world. [Sources: VETS press release, “Texas Launches Largest Publicly-Funded Psychedelic Research Initiative in History with $50 Million Investment in Ibogaine,” May 22, 2025; Texas Tribune, “With Rick Perry’s backing and $50 million from the state, Texas set to become a leader in psychedelics research,” June 11, 2025]
In December 2025, the Texas Health and Human Services Commission selected UTHealth Houston, in collaboration with the University of Texas Medical Branch at Galveston, to lead a multi-institution consortium called IMPACT (Ibogaine Medicine for PTSD, Addiction, and Cognitive Trauma). The consortium includes Texas Tech University, Texas Tech University Health Sciences Center El Paso, UT Austin, UT Health Science Center at San Antonio, UT Rio Grande Valley, UT Tyler, Texas A&M University, the University of North Texas Health Science Center, Baylor College of Medicine, and JPS Health Network.
Then, on March 31, 2026, eighteen days before the federal EO was signed, Lt. Gov. Dan Patrick and House Speaker Dustin Burrows announced that Texas would proceed with the program without a private drug company partner. Multiple proposals from drug companies had failed to meet the state’s eligibility standards.
The bar Texas had set was high. To qualify, a drug company had to commit to four things: a credible plan to obtain FDA approval for ibogaine, a corporate presence in Texas, a $50 million private match to the state’s $50 million, and 20% of any future revenue from an approved product going back to Texas. Rice University drug policy fellow Katharine Neill Harris, who tracks state psychedelic policy closely, said the conditions were “demanding” enough that it was “believable that prospective applicants did not meet the requirements.”
That left the state with a structural problem heading into April 2026. As Harris put it bluntly: “As SB 2308 is written, HHSC should not be able to release the $50 million in state funds to the consortium without the private match.”
The federal $50 million and the Texas gap
This is where the timing of the federal Executive Order, and the size of the federal allocation, become difficult to read as a coincidence.
There has been informal speculation across the field, though no official confirmation from either Texas state officials or the White House, that the federal $50 million through ARPA-H may be designed in part to backfill the private-sector match Texas was unable to secure. The numbers align exactly. The structure of Section 3, federal funding to support state-level programs, maps cleanly onto the Texas matching requirement. And the Texas Tribune updated its March 31 article on April 19, the day after the EO was signed, to add this line:
“This effort will also now get a federal boost, as President Donald Trump signed an executive order on April 18 to facilitate research into psychedelic drugs, including ibogaine, as a way to treat serious mental health illnesses. In particular, the order includes at least $50 million to support investments from states like Texas on this issue.”
Whether ARPA-H funds will legally qualify as the “non-state matching funds” SB 2308 requires, or whether Texas will need a legislative or administrative fix to accept them in that role, has not been publicly resolved. Harris noted that Texas “might need to make some legislative or legal fixes to meet the statutory requirements of SB 2308” if the state proceeds without a private drug-company partner.
A second caveat. Even with both $50 million pots combined, Texas would still face a substantial funding gap relative to the cost of a full FDA-approval drug development program. Harris estimated that “a drug development effort like this will likely require much more than $100 million overall, not just the initial $50 million in state funding.”
The bigger pattern: a multi-state ibogaine network
Texas is the most advanced state initiative, but it is no longer the only one. The federal EO arrives into a landscape where state-level momentum was already converging on Texas as a model.
Mississippi signed House Bill 314 into law on March 26, 2026, with Governor Tate Reeves’s signature. The Mississippi bill explicitly directs its research consortium to coordinate with and use the same drug developer as the lead consortium in another state, language widely understood to anchor Mississippi to the Texas framework.
Kentucky’s $21 million ibogaine research fund cleared key legislative hurdles in March 2026. West Virginia’s ibogaine bill (HB 4626) passed both chambers and was sent to the Governor on March 31, where it was subsequently vetoed. Bills are pending or advancing in Oklahoma, Tennessee, Missouri, Louisiana, Colorado, Georgia, Maryland, New Hampshire, New York, and Vermont. Utah passed legislation on March 4, 2026, authorizing a state mental health institute to conduct a clinical study on psychedelic-assisted therapy for veterans with treatment-resistant PTSD.
Trigger laws, which would automatically reschedule psychedelic substances at the state level if the federal government does so first, are advancing in South Dakota, Mississippi, and West Virginia. [Source: High Times, April 2026]
If implementation goes well, the result will be a federal-state research network operating across a dozen or more jurisdictions, sharing protocols and data, with Texas’s IMPACT consortium as the operational hub. The federal $50 million through ARPA-H sits as the connective tissue between these state initiatives and the FDA approval pathway that Sec. 2 and Sec. 5 of the Order are designed to accelerate.
If implementation goes poorly, Texas may face an unresolved legal question about its private-match requirement, other states may struggle to coordinate without a clear federal framework, and the matching funds may end up flowing into less well-developed state programs that cannot absorb them productively.
The mechanism is the most concrete near-term lever in the Order. The execution is what will determine whether it produces patient-relevant clinical activity in the next 24 months.
The institutional infrastructure to execute this well already exists across a handful of organizations focused on the policy and operational questions psychedelic medicine raises at the federal-state interface. The Ibogaine Healthcare Policy Institute (IHPI) is one of them.
2.4 HHS, FDA, VA, and private sector collaboration
The fourth mechanism addresses one of the most persistent structural problems in psychedelic medicine research: federal agencies have been running parallel work for years without systematically sharing what they are learning.
Section 4 of the Order reads: “The Department of Health and Human Services (HHS) and FDA shall collaborate with the Department of Veterans Affairs (VA) and, as appropriate and consistent with applicable law, including any privacy restrictions from the Privacy Act of 1974 and the Health Insurance Portability and Accountability Act of 1996, with the private sector, to increase clinical trial participation, data sharing, and real-world evidence generation regarding psychedelic drugs, and shall prioritize drugs that have received a Breakthrough Therapy designation.”
The Order then directs HHS, FDA, and the VA to sign data-sharing memoranda to ensure that clinical study data generated by other federal agencies is made available to the FDA to facilitate timely evaluation and approval under section 505 of the Federal Food, Drug, and Cosmetic Act.
Two elements in this provision deserve attention: the specific role assigned to the VA and the broader inter-agency data-sharing architecture it establishes.
The VA’s role
The Department of Veterans Affairs operates the largest integrated health care system in the United States. It is also the agency with the most direct exposure to the patient populations the Order is designed to serve: veterans with treatment-resistant PTSD, depression, and substance use disorder.
For most of the last sixty years, the VA maintained a careful distance from psychedelic research. That posture began to shift in late 2023.
In November 2023, VA leadership testified before the House Veterans Affairs Subcommittee on Health on emerging psychedelic therapies. In January 2024, the VA’s Office of Research and Development issued its first Request for Applications focused on MDMA or psilocybin for PTSD and depression. In April 2024, the VA chartered an Integrated Project Team of experts from across the agency to develop a comprehensive plan for psychedelic-assisted therapy implementation.
In December 2024, the VA announced its first directly-funded study of psychedelic-assisted therapy for veterans: an MDMA-assisted therapy program for PTSD and alcohol use disorder, funded at approximately $1.5 million over five years, conducted at the Providence VA Medical Center in Rhode Island and the West Haven VA Medical Center in Connecticut. It was the first time in nearly six decades that the VA had directly supported psychedelic research.
In November 2025, the VA confirmed that it was expanding its psychedelic-assisted therapy trials to nine facilities, including sites in the Bronx, Los Angeles, Omaha, Palo Alto, Portland (Oregon), and San Antonio. The trials evaluate MDMA and psilocybin alongside structured psychotherapy for veterans with treatment-resistant PTSD, depression, and anxiety disorders.
The Order now directs the VA to formalize and expand this work in coordination with HHS and FDA, and to share its data through memoranda of understanding designed to feed FDA’s evaluation pipeline. Congressional interest in scaling this work further is also active: the bipartisan Innovative Therapies Centers of Excellence Act, introduced in 2025 by Representatives Lou Correa and Jack Bergman (co-chairs of the Psychedelics Advancing Therapies Caucus) and Senator Ruben Gallego, would designate at least five VA medical facilities as innovative therapies centers of excellence with expanded research mandates.
Inter-agency data sharing
The data-sharing piece is structurally significant for a different reason. Psychedelic medicine research has been generated across a fragmented set of institutional actors: academic medical centers, the VA, NIH, NIDA, ARPA-H, private developers, and now state-funded consortia. Those data streams have historically not connected back to the FDA’s evaluation pipeline in any systematic way.
The Order directs that to change. If the data-sharing memoranda are signed and operate as intended, the FDA will, for the first time, have systematic access to clinical evidence generated outside its normal evaluation channels, with a specific priority on Breakthrough Therapy candidates.
The privacy carve-outs in the Order are doing real work in the text. The Order explicitly references the Privacy Act of 1974 and HIPAA. Patient-level data has historically been one of the harder elements of inter-agency coordination, and the Order does not override existing privacy law. What it does is direct the agencies to find arrangements that respect those limits while still moving data toward FDA evaluation.
Whether the agencies actually execute on this is a separate question. Inter-agency memoranda of the kind the Order contemplates have been written before for other purposes and have varied widely in how operational they ultimately prove to be.
2.5 The timely-rescheduling pathway
The fifth mechanism is the longest-horizon provision in the Order, and the one most likely to be misunderstood. It is also the section where the underlying pharmaceutical pipeline matters most.
Section 5 reads: “The Attorney General shall, in consultation with HHS, initiate and complete review of any product containing a Schedule I substance that has successfully completed Phase 3 clinical trials for a serious mental health disorder, so that rescheduling, if appropriate under 21 U.S.C. 811, may proceed as quickly as practicable for such specific products that are ultimately approved under section 505 of the Federal Food, Drug, and Cosmetic Act.”
In plain English: when a Schedule I psychedelic completes Phase 3 trials successfully and obtains FDA approval, the federal government commits to rescheduling it on the fastest practicable timeline.
How rescheduling actually works
The Controlled Substances Act provides the legal framework for rescheduling under 21 U.S.C. 811. The process can be initiated in three ways: by the Attorney General on the AG’s own initiative, by a request from the Secretary of HHS, or by a petition from any interested party.
Once initiated, HHS conducts a scientific and medical evaluation through the FDA, generally referred to as the Eight-Factor Analysis, and submits a recommendation to the Drug Enforcement Administration. The HHS findings on the scientific and medical questions are binding on DEA, until DEA initiates formal rulemaking. After that point, DEA must continue to give the HHS findings significant deference, and holds the final legal determination.
The marijuana rescheduling process offers a useful timeline reference. HHS submitted its recommendation to reschedule marijuana to Schedule III in August 2023. The DEA published its proposed rule in May 2024. As of early 2026, the formal rescheduling has still not been completed.
Section 5 of the Order is, in effect, a directive from the President to the Attorney General to compress that timeline for psychedelic candidates that complete Phase 3 trials. It does not change the underlying CSA process. The AG’s rescheduling authority is statutory and is exercised through a formal rulemaking procedure that a presidential directive can prioritize but cannot substitute for. The Order commits the administration to running that process quickly when the trigger conditions are met.
What the pipeline actually looks like
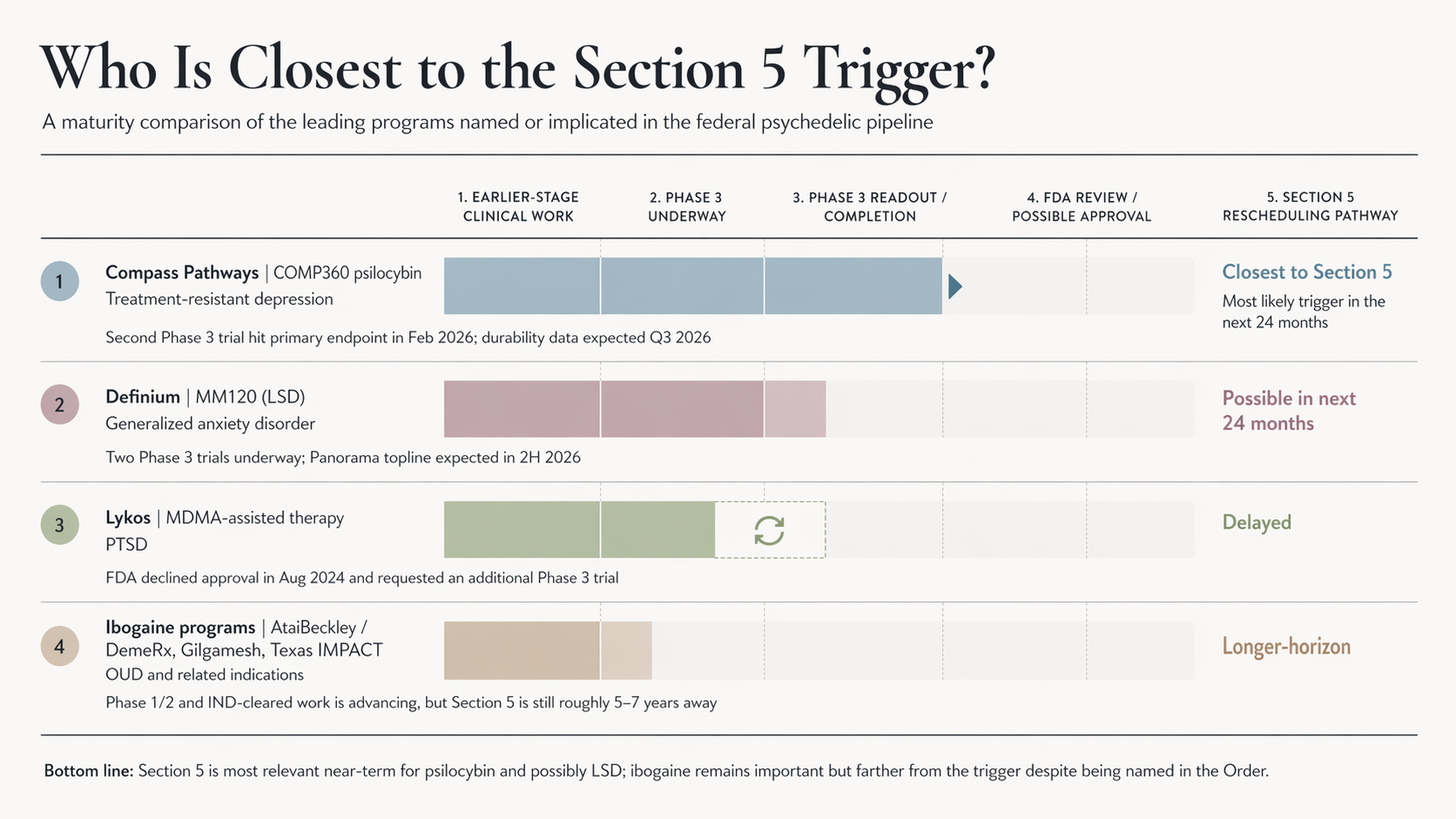
Section 5 only matters to the extent that there are psychedelic candidates approaching Phase 3 completion. There are several worth knowing.
Compass Pathways’ COMP360 (psilocybin) for treatment-resistant depression is the most advanced. Compass announced in February 2026 that COMP360 had successfully achieved its primary endpoint in a second Phase 3 trial. The 26-week durability data from the COMP006 trial is expected in early Q3 2026. This is currently the single most likely psychedelic candidate to trigger Section 5’s rescheduling pathway in the next 24 months.
Definium Therapeutics’ (MindMed) MM120 (lysergide D-tartrate, an LSD formulation) for generalized anxiety disorder has two Phase 3 trials underway: Voyage (US-only) and Panorama (US and European sites, dosed first patient in early 2025). Topline results from Panorama’s 12-week double-blind period are expected in the second half of 2026. Definium is also running a third Phase 3 trial, Emerge, evaluating MM120 for major depressive disorder.
GH Research’s GH001 (5-MeO-DMT) for treatment-resistant depression had its FDA clinical hold lifted in January 2026, after a multi-year process to address the agency’s regulatory requirements. GH Research now plans to initiate its Phase 3 program in the second half of 2026.
Helus Pharma’s (Cybin) CYB003 (a deuterated psilocybin analog) for major depressive disorder holds Breakthrough Therapy designation as an adjunctive treatment and is in late-stage clinical development.
MDMA-assisted therapy for PTSD sits in a more uncertain position. Lykos Therapeutics submitted an FDA New Drug Application based on its Phase 3 program, but the FDA declined approval in August 2024, requesting an additional Phase 3 trial. The path from here to Section 5 eligibility for MDMA is therefore longer than it appeared two years ago.
Ibogaine candidates are the most distant from Phase 3 completion, despite being explicitly named in the Order. Atai Life Sciences (now AtaiBeckley), through its DemeRx subsidiary, is advancing DMX-1002 (ibogaine HCl) for opioid use disorder through a Phase 1/2a trial, primarily in the UK. AtaiBeckley also holds DemeRx’s noribogaine candidate (DMX-1001). Gilgamesh Pharmaceuticals received a $19 million NIDA grant in 2024 to develop GM-3009, a “safer ibogaine analog” with reduced cardiotoxicity. On the day of the EO signing, FDA Commissioner Marty Makary announced IND clearance for ibogaine, signaling the FDA’s broader willingness to authorize US-based ibogaine clinical trials. That is a meaningful regulatory shift. It does not change the underlying timeline to Phase 3 completion. The Texas IMPACT consortium is now positioned to advance the underlying clinical work, but a US-based ibogaine candidate that triggers Section 5 realistically has a five-to-seven-year horizon, not a one-to-two-year one.
The implication
For the next 24 months, Section 5’s rescheduling pathway is mostly relevant to psilocybin (Compass) and possibly LSD (Definium). Either successful approval would be the first FDA-approved Schedule I psychedelic to be rescheduled under federal law in modern history. The political and regulatory significance of that first approval would be substantial, regardless of which compound gets there first.
For ibogaine specifically, Section 5 is a longer-horizon commitment. The near-term mechanisms in the Order, particularly the Right to Try pathway in Section 2(b) and the state-level matching funds in Section 3, are the provisions most likely to materially affect ibogaine patient access in the meantime.
Section 3: What the EO Does Not Do
A clear-eyed reading of any executive order has to include what it does not change. This section walks through the boundaries of the April 18 Order. None of these limits make the Order unimportant. Each one is worth understanding before forming an expectation about what comes next.
It does not reschedule any substance
Ibogaine, psilocybin, MDMA, and LSD all remain Schedule I controlled substances under the Controlled Substances Act. The Order commits the Attorney General to fast-track future rescheduling reviews when products complete Phase 3 trials and obtain FDA approval (Sec. 5), and no rescheduling action took effect on the day the Order was signed.
The most useful comparison point is the December 18, 2025 Executive Order on medical marijuana research, which directed the Attorney General to “take all necessary steps” to complete the rulemaking process to move marijuana from Schedule I to Schedule III. As of April 2026, that rescheduling has still not been finalized. HHS submitted its formal recommendation to reschedule marijuana in August 2023. The DEA published its proposed rule in May 2024. The December 2025 Executive Order added presidential pressure on top of that pre-existing administrative process. The rulemaking remains incomplete sixteen months after the proposed rule was published, and four months after the EO directed its completion.
That is the realistic timeline frame for any rescheduling action the April 18 Order may eventually trigger.
It does not create new appropriations
The $50 million allocated to ARPA-H under Sec. 3 is, in the Order’s own words, “from existing funds.” This is a reallocation directive. It tells HHS to find $50 million within the resources ARPA-H already has and to direct that money toward state-level psychedelic research programs.
ARPA-H operates on an annual budget of roughly $1.5 to $2.5 billion. A $50 million carve-out is meaningful and represents a fraction of the agency’s total resources. If those resources are reduced through subsequent budget actions, or if the carve-out comes at the cost of other ARPA-H priorities, the practical effect of Sec. 3 weakens.
It does not bind agencies in legally enforceable ways
Section 6(c) of the Order is explicit: the Order “is not intended to, and does not, create any right or benefit, substantive or procedural, enforceable at law or in equity by any party against the United States, its departments, agencies, or entities, its officers, employees, or agents, or any other person.”
In plain terms: if the FDA, the DEA, or the VA chooses to slow-walk implementation, no patient, physician, or developer has standing to sue under the Order itself. The Order is a directive within the executive branch. Its operational force depends on the willingness of the relevant agencies to execute on it.
The same Sec. 6(c) language appears in the December 2025 cannabis Executive Order. The slow pace of marijuana rescheduling in the months since is a useful illustration of what this limit looks like in practice.
It does not change FDA’s clinical evidence standards
A drug must still demonstrate safety and efficacy through the standard trial process. The Priority Voucher mechanism in Sec. 2(a) compresses the FDA review timeline. The bar for approval is unchanged.
This matters for managing expectations. Some psychedelic candidates will succeed in their Phase 3 programs and reach approval. Others will not. The August 2024 FDA decision on Lykos Therapeutics’ MDMA-assisted therapy application, which required an additional Phase 3 trial rather than approval, is recent evidence that the FDA’s evidence bar has held even as the political climate has shifted.
It does not address supply
No US developer currently holds a federally authorized domestic supply of pharmaceutical-grade ibogaine. Ibogaine reaching clinics and researchers today is sourced through two botanical pathways: direct extraction from Tabernanthe iboga, native to Gabon and culturally associated with Bwiti spiritual traditions, and semi-synthesis from voacangine extracted from Voacanga africana, a separate West and Central African plant. Most contemporary clinical and therapeutic supply is produced through the voacangine pathway. Fully synthetic and biosynthetic routes are in development but not yet operational at scale.
Gabon has been actively developing a legal export framework under the Nagoya Protocol, an international treaty governing access to genetic resources and equitable benefit-sharing with the communities and countries from which those resources originate. The first Nagoya Protocol-compliant shipment of iboga was authorized by the Gabonese government in 2023. Gabon also asserts, and Nagoya norms support, that benefit-sharing obligations extend to non-iboga sources of ibogaine, including voacangine-based semi-synthesis, on the grounds that the underlying therapeutic tradition originates with the Bwiti.
The Order does not address importation pathways, domestic manufacturing, or the international benefit-sharing structures that any legitimate US ibogaine supply will need to engage with. A future rescheduling event under Sec. 5 would create demand for FDA-approved ibogaine compounds that the existing US supply infrastructure is not currently equipped to meet.
It does not supplant Congress
Permanent rescheduling of any psychedelic compound, structural reform of how Schedule I research is conducted, and durable expansion of patient access through statute all require legislative action.
Congress has made some progress here. The HALT Fentanyl Act, signed by President Trump on July 16, 2025, included significant reforms to Schedule I research procedures. It established a new alternative registration process for certain Schedule I research and reduced several barriers to initiating studies on Schedule I controlled substances, including psychedelics.
Implementation of those provisions has been mixed. The Attorney General was required to issue research guidelines under the HALT Fentanyl Act by January 2026, and as of early 2026, that deadline had been missed. Other reform proposals remain pending, including the Veterans Health Administration Novel Therapeutics Preparedness Act (S.4220), introduced in March 2026 by Senators Tim Sheehy, Tammy Duckworth, Ruben Gallego, and John Boozman, which would establish a dedicated Office of Novel Therapeutics within the VA to oversee governance, workforce readiness, and clinical implementation infrastructure for emerging mental health therapies.
Two earlier vehicles, the Freedom to Heal Act and the Innovative Therapies Centers of Excellence Act, also remain pending, though the Novel Therapeutics Preparedness Act appears to have the strongest near-term path.
A future administration can revoke an Executive Order. Real durability for psychedelic medicine policy depends on Congressional action. The April 18 Order accelerates federal action within the executive branch’s authority, and that is the limit of what it can do on its own.
These limits are worth holding alongside the mechanisms walked through earlier. Together, they describe what the Order is: a substantive federal acceleration directive operating within real legal and political constraints. They are also the place where credible expectations begin, for the next 24 months and beyond.
Section 4: Why This Matters — The Underlying Need
The Executive Order is built around a population. Read alongside its accompanying Fact Sheet, the Order names veterans, Americans with treatment-resistant mental illness, and Americans struggling with substance use disorder as the people the policy is designed to serve. Each of those populations carries an underlying need that explains why a federal action of this scope was proposed and why the response across the political spectrum has been broadly favorable.
This section walks through what is actually at stake.
4.1 Veteran mental health
The numbers from the Order itself are sobering. More than 6,000 American veterans die by suicide every year, and that figure has held above 6,000 for over two decades. The veteran suicide rate is more than double the rate among non-veteran adults.
Veterans in their first year of separation from active service are at the highest risk, with suicide rates roughly twice the broader veteran rate during that twelve-month window. This was the specific population that the President’s first-term Executive Order 13822, signed in January 2018, was designed to reach. That order directed the VA, Department of Defense, and Department of Homeland Security to provide seamless mental health care access during the year following discharge.
The persistence of the underlying numbers since then, despite that earlier order and the broader investment in veteran mental health services, points to a deeper problem. For many veterans whose conditions are rooted in traumatic brain injury, blast exposure, or complex polytrauma, standard psychiatric treatments produce partial relief at best.
A meaningful number of veterans have stopped waiting for the system to catch up. Over the past decade, hundreds have traveled to clinics in Mexico, Costa Rica, and elsewhere to access ibogaine treatment that remains illegal at home. They have done this without insurance coverage, often at significant personal cost, and often after multiple cycles of conventional treatment have failed to help.
The clinical evidence base behind that underground movement has grown substantially. In January 2024, researchers at Stanford Medicine, in collaboration with Veterans Exploring Treatment Solutions (VETS), published the first prospective observational study of magnesium-ibogaine treatment in 30 US Special Operations Forces veterans with traumatic brain injury. The study, published in Nature Medicine, documented large and sustained improvements in PTSD symptoms, depression, anxiety, suicidal ideation, and overall functioning following a single ibogaine treatment combined with a cardioprotective magnesium protocol. The cohort had independently scheduled themselves for treatment at a clinic in Mexico, and the Stanford team conducted structured pre- and post-treatment assessments.
A follow-up Stanford analysis, published in Nature Mental Health in July 2025, used neuroimaging to identify specific neurophysiological signatures of recovery, including changes in theta brain wave activity associated with cognitive flexibility and reductions in cortical complexity associated with PTSD remission.
Earlier observational work by The Mission Within and collaborators, published in Chronic Stress in 2020, documented similar improvements in PTSD, depression, and cognitive function across a cohort of Special Operations Forces veterans treated with ibogaine and 5-MeO-DMT in Mexico.
At the April 18 signing ceremony, former Navy SEAL Robert O’Neill described his own ibogaine treatment as having “fixed my demonic relationship with alcohol” and as having saved his life. His testimony reflected a broader pattern that the published research is now beginning to substantiate: a meaningful number of veterans who have accessed ibogaine outside the United States describe outcomes that go beyond what conventional treatment had been able to deliver for them.
4.2 Opioid use disorder and treatment-resistant addiction
The opioid crisis is the second underlying need the Order is responding to.
In 2024, US drug overdose deaths fell by approximately 27% compared to 2023, the largest single-year decline since the crisis began. Opioid-involved deaths dropped from approximately 83,000 in 2023 to roughly 54,000 in 2024. Provisional CDC data from early 2026 suggest the decline continued through 2025.
That progress is real and meaningful. It is also incomplete. Drug overdose remains the leading cause of death for Americans aged 18 to 44. The 2024 decline brought opioid deaths only modestly above pre-pandemic 2019 levels. Progress has been uneven across states, with most seeing substantial declines and several still reporting rising deaths.
Standard medication-assisted treatment using buprenorphine, methadone, or naltrexone is the current evidence-based first line for opioid use disorder, and its expanded availability is one of the factors driving the 2024 decline. These medications work for many patients. They do not work for everyone. A substantial fraction of patients cycle through repeated relapses and remain at high risk of fatal overdose despite engagement with treatment.
Ibogaine has been studied in opioid use disorder for several decades, primarily through observational and small clinical studies conducted outside the United States. A 2018 twelve-month follow-up observational study by Noller and colleagues, published in the American Journal of Drug and Alcohol Abuse, documented sustained reductions in opioid use in patients treated with a single ibogaine session. A 2018 paper by Mash and colleagues in Frontiers in Pharmacology described ibogaine’s capacity to transition opioid-dependent patients from active use to abstinence with a significant reduction in withdrawal symptoms. A 2023 systematic review in Current Neuropharmacology summarized the broader ibogaine and noribogaine evidence base in substance use disorders.
Pharmacologically, ibogaine acts on multiple neurotransmitter systems simultaneously, including serotonin, dopamine, opioid, and NMDA receptors. Its mechanism is structurally different from the opioid agonist and antagonist mechanisms of standard medication-assisted treatment. That mechanistic distinction is part of why ibogaine has shown signal in patients for whom standard treatments have not worked.
The clinical concern with ibogaine is real and worth naming. Ibogaine has been associated with cardiac arrhythmia, including QT interval prolongation, and isolated cases of fatal cardiac events. The Stanford magnesium-ibogaine protocol was designed specifically to address this concern through the cardioprotective coadministration of magnesium and ibogaine. Parallel research efforts, including Gilgamesh Pharmaceuticals’ work on a “safer ibogaine analog” (GM-3009), are pursuing derivative compounds with reduced cardiac risk.
4.3 The broader serious mental illness picture
The Order’s framing of “serious mental illness” reaches beyond opioid use disorder and PTSD. More than 14 million American adults live with a serious mental illness, and roughly 8 million rely on prescription medication for these conditions. For a meaningful share, those medications produce only partial relief.
This broader population has driven the expansion of psychedelic clinical research over the past two decades. Compass Pathways’ COMP360 psilocybin program for treatment-resistant depression, with two successful Phase 3 trials announced as of February 2026, is the most advanced. Definium’s MM120 program for generalized anxiety disorder, with three Phase 3 trials underway, is close behind. MDMA-assisted therapy for PTSD remains in clinical development following the FDA’s August 2024 decision not to approve Lykos Therapeutics’ (now Resilient Pharmaceuticals’) application without an additional Phase 3 trial.
The thread shared across these programs and the underlying need the Executive Order addresses is the limit of what conventional treatments have been able to do for patients who need them most. The Order does not commit to any particular outcome. It commits to faster federal action to determine what these treatments can and cannot deliver.
Section 5: The State-Level Story
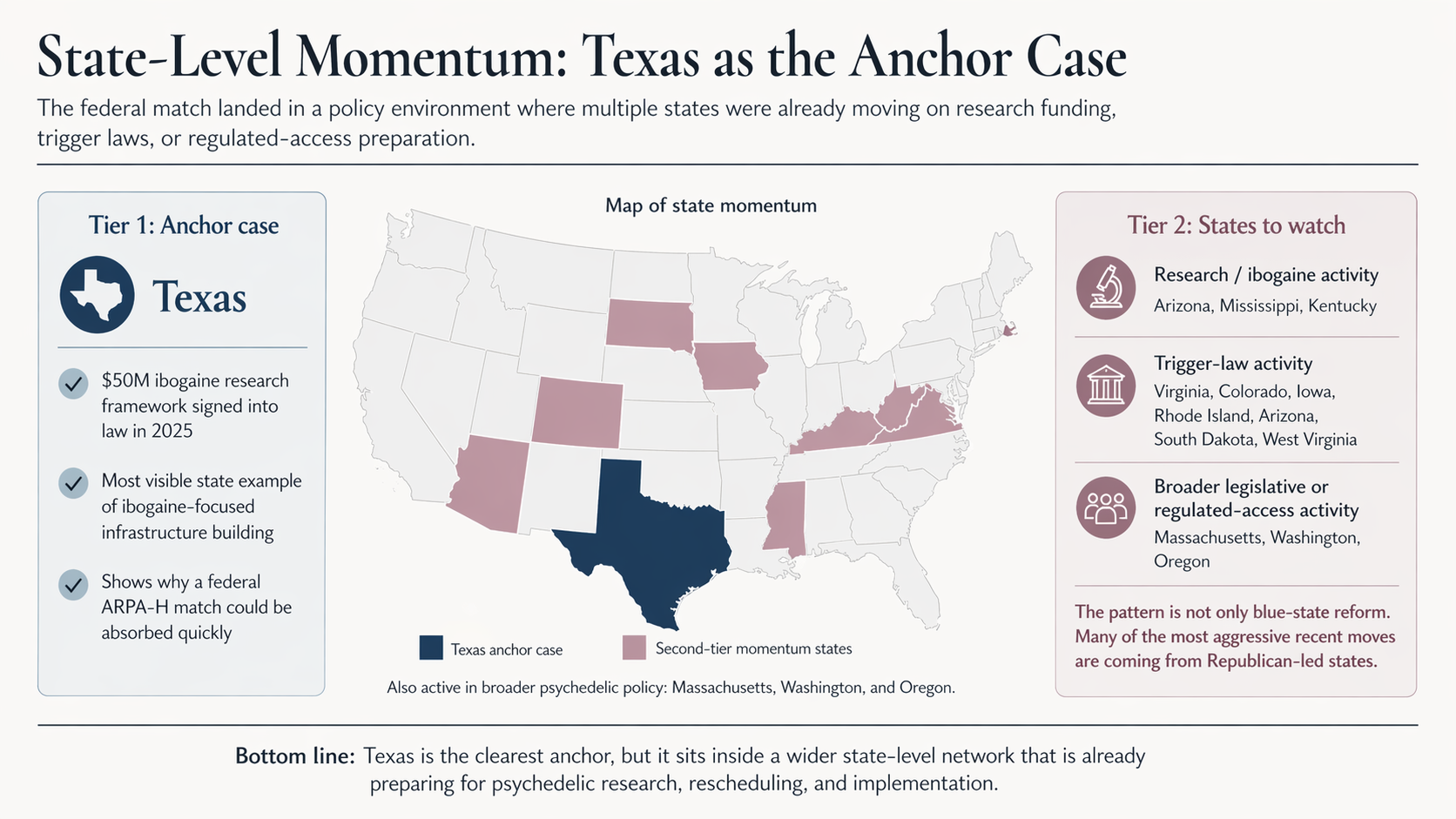
Federal psychedelic policy has moved slowly. State psychedelic policy has not. By the time President Trump signed the Executive Order on April 18, more than a dozen US states had passed, considered, or enacted laws creating space for psychedelic research, regulated access, or post-approval rescheduling. Texas’s $50 million ibogaine research consortium is the most visible example. It is far from the only one. The story behind that activity is worth understanding because it explains why a $50 million federal match landed in a policy environment already structured to absorb it.
5.1 The Kentucky story
The most consequential figure in the recent push for state-level ibogaine research is not, at the moment, in Texas. He is in Kentucky.
In June 2022, Kentucky Attorney General Daniel Cameron appointed W. Bryan Hubbard, a Republican lawyer with a long career in state government, to chair the Kentucky Opioid Abatement Advisory Commission. The Commission’s job was to allocate the state’s $842 million share of national opioid settlements to programs designed to address an epidemic that had devastated Kentucky’s working-class communities for two decades.
In the summer of 2022, Hubbard learned about ibogaine. By January 2023, he was describing it to Cameron as a “Manhattan Project opportunity” for the state. By May 2023, the Commission announced its intention to allocate $42 million from the settlement, paired with matching investment from a drug developer, to fund what would have been the first publicly funded clinical trial of ibogaine for opioid use disorder in the United States.
The proposal did not survive Kentucky’s 2023 election cycle. Cameron ran unsuccessfully for governor against incumbent Andy Beshear that November. When Cameron’s term as attorney general ended at the close of 2023, the incoming attorney general, Russell Coleman, moved to replace Hubbard as chair of the Commission. Hubbard left in mid-December 2023, and the Kentucky Ibogaine Initiative collapsed shortly after.
Hubbard kept moving. He took the proposal to Ohio and South Dakota, where it was deemed too outlandish for strapped state budgets, and finally to Texas, where the nonprofit Texans for Greater Mental Health brought him into a state heading into a $16 billion budget surplus. In June 2025, he became the inaugural CEO of Americans for Ibogaine, a national advocacy nonprofit dedicated to public education and policy reform around ibogaine for substance use disorder, traumatic brain injury, and co-occurring mental health conditions. Texas’s $50 million ibogaine research framework, signed into law that same month, advanced a structurally similar concept to Hubbard’s Kentucky proposal in a state where the political conditions proved more favorable.
The Hubbard story matters because it shows what state-level activity actually looks like up close. Most of the work is done by individuals operating within state government, with narrow procedural windows, persuading attorneys general, legislators, and budget directors to allocate funds for treatments that remain federally illegal.
5.2 Why this is mostly Republican territory
The most striking feature of recent state-level psychedelic activity is which states are leading. Texas, Mississippi, Kentucky, Utah, Arizona, West Virginia, Tennessee, Oklahoma. These are not states where progressive drug-policy reform typically begins. The conventional path of psychedelic policy reform, beginning with Oregon’s Measure 109 in 2020 and Colorado’s Proposition 122 in 2022, looked like a continuation of the cannabis playbook. Blue-state coastal voters approve a ballot measure. Other blue states follow. Federal action eventually catches up.
The current wave is structured differently. It is happening through state legislatures rather than at the ballot box. It is being led by Republican lawmakers and conservative-aligned advocacy organizations. The framing emphasizes veteran mental health, opioid recovery, and economic opportunity rather than recreational liberalization or broader drug-policy reform.
That framing has political weight in places where the underlying need is most acute. Kentucky, West Virginia, Ohio, and the rural counties of Texas, Mississippi, and Tennessee have borne some of the heaviest per-capita opioid overdose burdens in the country for two decades. Veterans constitute a politically influential constituency in many of these same states, with rural veteran density higher than the national average across the South and Mountain West. The psychedelic-medicine pitch, when delivered as a treatment-of-last-resort for opioid use disorder and combat-related PTSD, looks very different from a recreational legalization pitch.
Blue and purple states have not been absent from this picture. Massachusetts, Colorado, Washington, and Oregon all have active legislative or regulated-access activity, and the Berkeley law and policy map shows reform proposals in roughly half of all US states. The pattern is not red against blue. It is that the most aggressive recent moves on ibogaine specifically, framed around veterans and opioid use disorder, have come from states with Republican legislative majorities.
5.3 Trigger laws as a parallel track
Beyond the research-funding statutes that Texas, Mississippi, and (briefly) Kentucky have pursued, a second category of state legislation is moving in parallel: trigger laws.
A psychedelic trigger law schedules a specific substance at the state level the moment the federal government does, or, in some versions, the moment the FDA approves a drug containing it. Functionally, trigger laws are insurance against state-level regulatory delay. If the FDA approves Compass Pathways’ COMP360 psilocybin or Definium’s MM120 LSD-derived therapy, a state with a trigger law on the books would not need a separate legislative session to make the new medicine accessible to its patients.
Trigger law bills have been introduced or are advancing in Virginia, Colorado, Iowa, Rhode Island, Arizona, South Dakota, and West Virginia. Compass Pathways has publicly supported several of them. Arizona is a useful illustration of how the two tracks can layer: the legislature approved a rescheduling trigger bill in 2025 and earmarked $5 million in the state’s FY2026 budget to fund ibogaine studies, combining post-approval positioning and pre-approval research funding in the same budget cycle.
The trigger law category is worth tracking separately because it indicates where states think federal action is headed. A trigger law is, in effect, a state-level prediction that the FDA will approve a psychedelic medicine within the relevant legislative horizon. The growing number of trigger law bills suggests that legislators in a meaningful range of states now consider FDA approval of at least one psychedelic-assisted therapy a plausible near-term outcome. Section 5 of the Executive Order, which directs the Attorney General to initiate expedited DEA rescheduling reviews after FDA approval, is the federal-level mirror of that prediction.
5.4 What the network adds up to
Taken together, the state-level activity describes a quiet form of parallel federalism. Texas, Mississippi, Kentucky (now reportedly developing a separate $21 million effort despite the 2023 collapse), Arizona, Utah, and the trigger-law states are not waiting for federal action. They are building research, funding, and regulatory infrastructure in advance of it.
The Executive Order’s $50 million federal match changes the math for these states. Fifty million dollars matched against $42 million in state contributions, or $5 million, or some combination from a half-dozen states, is not a sum that will fund a national program. It is a sum that signals federal alignment with the states already in motion. The signal matters more than the dollar figure because it lowers the political risk for the next state legislator considering an ibogaine research appropriation and signals to in-motion states that their direction of travel is shared.
The complication is that state research dollars and trigger laws sit on opposite sides of the regulatory wall. Research dollars fund work that does not require federal approval to begin, since clinical trials operate under FDA-authorized investigational new drug protocols. Trigger laws position states to respond after FDA approval. The space in between, where coverage decisions, payer policy, clinical guidelines, delivery infrastructure, and quality assurance get built, is largely the work the Executive Order leaves untouched.
That space matters because of what it determines. A new medicine that is FDA-approved but not covered by Medicaid or VA, not reimbursed by commercial payers, not delivered through accredited clinical settings, and not standardized across clinical practice does not reach the patients the EO is supposed to serve. State research consortia produce evidence. Trigger laws produce regulatory clearance. Neither produces the implementation pathway between approval and access. That is the work of the next phase, and it is where institutes, state agencies, payer organizations, and provider networks will have to do most of the lift.
Section 6: What Access Actually Looks Like for Patients
For patients, families, and clinicians reading about the Executive Order, the most important question is the most practical one. What does any of this mean for someone who needs treatment now, or who might benefit from a psychedelic-assisted therapy in the future?
The honest answer is that the Order opens federal pathways without, by itself, creating immediate access. There are three pathways that matter for patients in the near and medium term: clinical trial enrollment, the Right to Try and expanded access mechanisms, and post-approval prescription access once the FDA approves a specific medicine. This section walks through each.

6.1 Clinical trial enrollment
For most US patients today, enrolling in a clinical trial remains the most realistic regulated pathway to access psychedelic-assisted therapy. Trials are recruiting across multiple compounds and conditions: Compass Pathways’ Phase 3 program for treatment-resistant depression, Definium’s MM120 trials for generalized anxiety disorder, GH Research’s GH001 program, Helus’ CYB003, and a smaller number of academic and government-funded studies through institutions like Johns Hopkins, NYU, and Stanford.
For ibogaine specifically, the picture is more constrained. Most ibogaine clinical research is currently conducted outside the United States. AtaiBeckley/DemeRx’s DMX-1002 program and Gilgamesh Pharmaceuticals’ GM-3009 program are advancing, but US-based ibogaine trials have been limited by the combination of Schedule I status and FDA caution about cardiac safety. The Texas IMPACT consortium and the federal data-sharing provisions in Section 4 of the Order are designed to expand US-based research capacity over the next several years. The FDA’s IND clearance for ibogaine, announced on the day of the EO signing, should open the door to additional US-based trial activity in the months ahead. At the time of writing, however, US-based ibogaine trial options remain limited.
The most reliable way for patients to find current trials is the federal ClinicalTrials.gov database, which lists all FDA-regulated trials open for enrollment in the United States. Eligibility criteria vary by trial and condition, and most psychedelic trials have substantial exclusion criteria around cardiac history, current medications, and prior psychiatric diagnoses.
6.2 Right to Try and expanded access
The Executive Order’s Section 2(b) directs the FDA and DEA to establish a pathway for eligible patients to access investigational psychedelic drugs, including ibogaine compounds, under the federal Right to Try Act. This is the provision that has received the most patient-focused attention.
To be eligible under Right to Try as currently written, a patient must have a life-threatening or serious illness, must have exhausted FDA-approved treatment options, and must be unable to participate in a relevant clinical trial. The drug they seek must have completed at least Phase 1 clinical testing. A treating physician must request access from the manufacturer, and the manufacturer must agree to provide the drug.
Two practical complications follow.
The first is the Schedule I problem. Until the EO, the DEA’s position was that the Schedule I status of substances like psilocybin and MDMA prevented physicians from administering them under Right to Try, even when the underlying eligibility criteria were met. The Ninth Circuit affirmed that position in February 2025 in a case brought by Dr. Sunil Aggarwal. Section 2(b) of the EO directs the DEA to fix this administratively, including through “any necessary Schedule I handling authorizations for treating physicians and researchers.” Whether the DEA delivers, and on what timeline, is one of the open implementation questions.
The second complication is more specific to ibogaine. Mason Marks of the Petrie-Flom Center at Harvard Law School has noted that ibogaine arguably does not meet the basic safety requirements that the Right to Try Act presupposes, given the limited Phase 1 research conducted in the United States and the documented cardiac concerns. The EO directs the agencies to build a Right to Try pathway for ibogaine, but a near-term ibogaine Right to Try program would likely require either accelerated US Phase 1 work, formal FDA acceptance of foreign Phase 1 data, or both. Some of that work is now underway through the IND clearances granted to Atai/DemeRx and Gilgamesh, though it has not yet generated the kind of completed Phase 1 evidence base that would meet the legal threshold.
A parallel and longer-established mechanism, FDA expanded access (also known as compassionate use), already exists for investigational drugs that have not yet received approval. “MAPS Public Benefit Corporation, which became Lykos Therapeutics and was subsequently renamed Resilient Pharmaceuticals, used the expanded access pathway to provide MDMA to a small number of patients during its Phase 3 program. Expanded access is administered by the FDA through a defined application process and may, in practice, be the more workable near-term route for patients seeking access to specific psychedelic compounds whose developers are willing to participate.
In every version of these pathways, the binding constraints sit downstream of the legal authority. A patient needs a treating physician willing to administer the drug, a manufacturer willing to provide it, a clinical setting equipped to manage the cardiac and psychiatric monitoring requirements, and the financial means to cover the treatment, since neither Right to Try nor expanded access requires any payer to cover the cost. The Order does not change any of these constraints.
6.3 Post-approval prescription access
The medium-term horizon, two to four years out for the most advanced candidates, is post-approval prescription access. If the FDA approves Compass Pathways’ COMP360 psilocybin for treatment-resistant depression, or Definium’s MM120 for generalized anxiety disorder, those medicines will become legally prescribable in the United States once the DEA completes the rescheduling that follows FDA approval. The EO’s Section 5 directs the Attorney General to expedite that rescheduling review, though, as the Petrie-Flom commentary notes, the DEA is already required by statute to reschedule within 90 days of FDA approval.
What post-approval access will actually look like depends almost entirely on the Risk Evaluation and Mitigation Strategy, or REMS, that the FDA attaches to any psychedelic approval. Glenn Cohen has framed the spectrum sharply. At one end is a delivery model that resembles ketamine clinics today: specialized providers, supervised sessions, significant out-of-pocket costs, limited insurance coverage, and concentrated geographic availability. At the other end is a model closer to prescribing fluoxetine or sertraline through a primary care provider, integrated into general practice, reimbursed through standard insurance pathways, and broadly available. The difference between those two endpoints will determine how many Americans actually receive these treatments.
For ibogaine specifically, post-approval access is a longer horizon. Based on where the leading candidates sit in the clinical pipeline, an FDA-approved ibogaine medicine is realistically a five-to-seven-year prospect, contingent on the Phase 2 and Phase 3 work that is only now beginning in earnest. The Order accelerates the federal posture toward that horizon. It does not collapse the timeline.
6.4 The honest near-term assessment
For most US patients, the Executive Order does not create new immediate access in 2026. It creates a more favorable federal posture toward access pathways that were already constrained by a combination of regulatory, clinical, and reimbursement realities.
Patients who may see meaningful changes within the next twelve to eighteen months are concentrated in three groups.
- Veterans may benefit from expanded VA research participation, particularly through the network of VA research sites added under the 2024 RFA process.
- Patients with treatment-resistant depression may have access to clinical trial slots at Compass Pathways and Definium sites, with the possibility of post-approval access following Compass’s expected late-2026 NDA filing / rolling NDA submission.
- Patients in states with active research consortia, beginning with Texas, may see additional trial capacity come online as those programs activate.
For everyone else, the practical near-term advice is unchanged from before the Order. Discuss psychedelic-assisted therapy with a treating clinician honestly. Search ClinicalTrials.gov for current enrollment opportunities. Be cautious about clinics, retreats, and providers operating outside the regulated pathways, where the safety, efficacy, and ethical standards are highly variable. Recognize that meaningful access for most conditions and most patients remains months to years away, and that the gap between FDA approval and actual access will be shaped by coverage and delivery decisions that the Executive Order does not address.
That gap is where the work of the next phase has to happen.
Section 7: What Remains Unresolved
The Executive Order is a meaningful federal action. It is also, like every executive order, a directive that depends on agencies, budgets, and political continuity to translate into substantive change. Several open questions will shape whether the Order amounts to a durable policy or remains a signaling document. The most consequential are worth naming directly.
7.1 Agency follow-through
The first question is whether the named agencies will actually act on the directives. The Order assigns specific responsibilities to the FDA, the DEA, the Department of Health and Human Services, the Department of Veterans Affairs, and the Attorney General. Each has its own institutional culture, statutory constraints, and capacity. The pace of implementation will vary across them.
The DEA is the agency to watch most closely. Section 2(b) directs the DEA to facilitate Schedule I handling authorizations for treating physicians and researchers under Right to Try, which is the administrative fix for the legal barrier that has prevented patient access since the Ninth Circuit’s Aggarwal ruling. The DEA has a long track record of moving slowly on Schedule I research authorizations. The HALT Fentanyl Act, signed in July 2025, included provisions designed to streamline Schedule I research registrations. As of April 2026, the Attorney General has missed the January 2026 statutory deadline for issuing the implementing guidelines required by that legislation. The same agency is now being asked to move quickly on a separate set of psychedelic-specific authorizations. Whether it does so will determine how quickly the Right to Try pathway becomes operational.
The FDA’s situation is more favorable. FDA Commissioner Marty Makary announced investigational new drug (IND) clearance for ibogaine on the same day as the EO signing, which means US-based ibogaine clinical trials can now formally proceed under FDA-authorized protocols. Combined with the priority voucher mechanism in Section 2a of the EO, the FDA pathway has more institutional momentum behind it than the DEA pathway. FDA has already acted on that mechanism by issuing three National Priority Vouchers on April 24, covering psilocybin for treatment-resistant depression, psilocybin for major depressive disorder, and methylone for PTSD. This makes the FDA follow-through the strongest early implementation signal in the Order so far.
7.2 The cannabis precedent
The most relevant comparison is one we have already discussed, and it is worth returning to. In December 2025, President Trump signed Executive Order 14370 directing the Attorney General to facilitate the rescheduling of marijuana from Schedule I to Schedule III. That order used the same Section 6(c) “subject to applicable law and the availability of appropriations” caveat as the April 2026 psychedelics order. As of April 2026, sixteen months after the HHS recommendation that initiated the process and four months after the President’s directive, the rescheduling has not been finalized. The DEA review remains ongoing.
The cannabis precedent does not predict that the psychedelics order will fail. It does establish that a presidential directive on Schedule I rescheduling, even one accompanied by political momentum, does not by itself produce regulatory change on a predictable timeline. The same dynamic is now in play for psychedelics, with the additional complication that the underlying drug development pipeline for psychedelics is still maturing.
7.3 Appropriations and budget durability
The $50 million federal match for state research is being drawn from existing ARPA-H funds, not from new appropriations. The Order’s text is explicit that the funds are to come “from existing funds.” This matters for two reasons.
The first is that ARPA-H, the Advanced Research Projects Agency for Health, is itself a relatively new institution. Created by Congress in 2022 with bipartisan support, ARPA-H operates on a discretionary budget that is contested in every appropriations cycle. The agency was chosen for the Order in part because of its operational nimbleness and willingness to fund unconventional research, including its $100 million EVIDENT program for clinical trial innovation launched in November 2025. Drawing $50 million from ARPA-H for state psychedelic matching is a meaningful share of the agency’s discretionary capacity. Whether subsequent budget cycles preserve that capacity, or whether ARPA-H itself faces budget pressure, will determine whether the matching mechanism continues beyond the initial allocation.
The second is that $50 million from existing funds is not the same as $50 million in new federal commitment. Reallocation does not signal sustained Congressional buy-in. A more durable version of this policy would be a line item in an HHS appropriations bill specifically dedicated to state-federal psychedelic research partnerships, ideally with multi-year authorization. That has not happened. Whether it happens in the FY2027 budget cycle is one of the most consequential questions for the durability of the Order’s funding mechanism.
7.4 The supply chain problem
Of all the unresolved questions, the supply chain question is the least discussed and may be the most binding. It is also specific enough to ibogaine that it needs to be named directly.
Ibogaine can be sourced through several distinct production pathways, and understanding which pathways are actually being used today is essential to understanding the policy problem. The botanical source most commonly associated with ibogaine is Tabernanthe iboga, a shrub native to Central Africa. Gabon is the primary range country for iboga, where the plant has a centuries-long history of ceremonial and medicinal use among Bwiti practitioners.
In 2019, the Gabonese government suspended iboga exports pending compliance with the Nagoya Protocol on Access and Benefit-Sharing, an international treaty governing the equitable use of genetic resources. In May 2023, after several years of negotiation involving the Gabonese Ministry of Forests, the nonprofit Blessings of the Forest, and the Vancouver-based Filament Health, the first Nagoya-compliant export of iboga left Gabon. Filament partnered with Terragnosis, founded by Jonathan Dickinson, to produce a purified total alkaloid extract. The first batch was directed to Ambio Life Sciences, an ibogaine clinic operator in Mexico.
That Ambio shipment remains the exception rather than the rule. According to a 2025 sourcing analysis by Dr. Joseph Barsuglia, most ibogaine used by clinics and research studies today is produced through a different pathway: chemical semi-synthesis from voacangine, an alkaloid extracted from Voacanga africana, an evergreen tree native to West and Central Africa. Voacanga africana contains voacangine at roughly 1.7% of root bark dry weight, approximately six times the ibogaine concentration of iboga root bark, and reaches harvestable maturity in three to five years compared to the seven-to-ten-year cycle for iboga. Most Mexican clinics and research sponsors rely on voacangine-to-ibogaine semi-synthesis rather than direct iboga extraction. Two additional pathways, full chemical synthesis from pyridine and biosynthesis through engineered yeast or bioreactors, are under development but not yet operational at scale.
The Voacanga pathway does not resolve the international-compliance problem. Gabon has asserted, and Nagoya Protocol norms support the view, that ethical sourcing of ibogaine, regardless of the botanical starting material, should involve reciprocity and benefit-sharing structures with Gabon as the Bwiti-tradition range country. This is the point Barsuglia’s analysis makes explicitly: “non-iboga sources still need Nagoya/reciprocity structure with Gabon.”
Rolling Stone’s April 2026 reporting on the Aspen Americans for Ibogaine conference captured the policy gap in stark terms. Bwiti grand master Me Moubeyi Bouale, present with a Gabonese delegation that included Ambassador Noël Nelson Messone and members of Blessings of the Forest, argued publicly for US accession to the Nagoya Protocol. Rick Perry, asked about Nagoya, told the reporter he had thought it was a Tex-Mex restaurant chain. The distance between those two positions, one articulating a centuries-old stewardship claim and the other operating with no awareness of the underlying international framework, is the distance the implementation work still has to cover.
Voacanga is not an end-run around Nagoya. It is a parallel source that still raises the same international-ethics questions, and any US clinical trial sponsor or approved-drug manufacturer will need to engage with those questions, whether sourcing from iboga, Voacanga, synthetic routes, or biosynthesis.
That milestone — the 2023 Filament-Ambio shipment — matters because it established a model for ethically sourced, internationally compliant iboga supply. It also exposed how thin the legitimate supply chain remains. As of early 2026, no equivalent Nagoya-compliant pipeline has been established for delivery to the United States. US importation of a Schedule I substance like ibogaine requires additional DEA importer licensing and per-shipment authorizations that sit on top of source-country compliance work. A US-based clinical trial sponsor with FDA approval and DEA researcher registration still needs a verified, importable supply.
The implications are concrete. Texas’s IMPACT consortium, Mississippi’s research bill, and any future ibogaine-specific Right to Try program all depend on the existence of a US-bound, compliant supply chain. None of that infrastructure currently exists at scale. Building it requires coordination between the Gabonese government, supply chain partners, the FDA, the DEA, and the funding entities, none of which the Executive Order directly addresses. A bipartisan working group on supply chain compliance, situated either in HHS or as an interagency body, would be one structural response. Whether anything like that materializes is open.
The further complication is that several entities are pursuing fully synthetic and semi-synthetic ibogaine analogs that aim to reduce or eliminate the botanical dependency entirely. AtaiBeckley/DemeRx’s DMX-1002 program and Gilgamesh’s GM-3009 program both work with synthesized molecules. If those programs receive FDA approval, dependence on Gabonese natural supplies may diminish, though the Nagoya reciprocity question may persist to the extent that the underlying chemistry is recognized as derived from traditional Gabonese medicine.
That outcome is several years away in any case. In the interim, the supply pipeline supporting most ibogaine clinical and therapeutic work remains fragile and concentrated in a small number of Voacanga-sourced semi-synthesis operations.
7.5 Cardiac safety and the ibogaine Phase 1 question
The cardiac safety profile of ibogaine, discussed in Section 4, intersects directly with the Phase 1 evidence question raised in Section 6. The FDA has historically required a more substantial Phase 1 safety package for ibogaine than for most other Schedule I substances, given the documented risk of QT prolongation and cardiac arrhythmia. The Stanford magnesium-ibogaine protocol is one approach to this concern, supported by publications in 2024 Nature Medicine and 2025 Nature Mental Health. Gilgamesh’s GM-3009 is another, oriented toward an ibogaine analog with reduced cardiac risk.
Neither resolution is yet field-wide. The FDA’s IND clearance for ibogaine, announced on the day of the EO signing, opens the door to US-based Phase 1 work, but generating a complete Phase 1 safety dataset for any specific ibogaine formulation will take time. Until that dataset exists, the Right to Try pathway for ibogaine remains legally and clinically constrained, even with the EO directive in place.
The FDA announced on April 24 that DemeRx NB may begin an early-phase U.S. clinical study of noribogaine hydrochloride, an ibogaine derivative, for alcohol use disorder following an IND submission. DemeRx describes the asset as DMX-1001, oral noribogaine, and says completion of the planned alcohol-interaction trial would support a Phase 2 start in 2027. This is important for ibogaine-adjacent development, but it is not one of the three voucher awards and does not bring ibogaine itself near Phase 3.
This is a case where the practical timeline is determined by clinical science rather than by federal policy. The Order can accelerate the regulatory environment around the science. It cannot accelerate the science itself.
7.6 Coverage, reimbursement, and delivery infrastructure
The most consequential gap, and the one that will determine whether any of this policy reaches patients at scale, is the gap between FDA approval and actual access. The Executive Order does not address coverage decisions by Medicare, Medicaid, the VA, or commercial payers. It does not address Risk Evaluation and Mitigation Strategy (REMS) requirements that the FDA will attach to any approval. It does not address clinical training, accreditation of delivery sites, or quality assurance for psychedelic-assisted therapy.
These omissions are not failures of the Order. They are simply outside its scope. Coverage policy is the work of CMS and private payers. REMS is the FDA’s work in consultation with sponsors. Clinical training and accreditation are the work of professional societies, state licensing boards, and credentialing bodies. The Order can create the upstream conditions that necessitate these decisions. It cannot make the decisions itself.
What this means in practice is that even in the most favorable implementation scenario, where the DEA acts quickly on Schedule I authorizations, where the FDA processes priority vouchers efficiently, where states stand up additional research consortia, and where one or more psychedelic medicines reach FDA approval within the next two to three years, the question of who actually gets treated will be settled by a different set of decisions made in different places by different actors. That work has not yet begun in earnest.
7.7 Political durability
The final unresolved question is political. The Executive Order is one of the administration’s actions. A subsequent administration could rescind it or decline to enforce it. The comparison to President Biden’s October 2022 marijuana directive is instructive. That directive launched an HHS scientific review, an HHS recommendation in 2023, a DOJ proposed rule in 2024, and a DEA review process that remains incomplete. Each step required sustained executive attention. Several stalled when attention shifted.
The bipartisan veteran framing of the psychedelics order offers some political insulation. Veteran mental health is one of the few federal policy areas where Republican and Democratic administrations have maintained broad continuity over the past decade, and the underlying constituency does not disappear with an election cycle. That insulation is partial, however. Permanent change requires Congressional action.
The closest existing legislative vehicle is the Veterans Health Administration Novel Therapeutics Preparedness Act (S.4220), introduced in March 2026 by Senators Sheehy, Duckworth, Gallego, and Boozman, which would establish an Office of Novel Therapeutics within the VA. Two earlier vehicles, the Innovative Therapies Centers of Excellence Act (Correa, Bergman, Gallego) and the Freedom to Heal Act (Booker, Paul, Dean, Mace), also remain pending. None has advanced to a floor vote. The Order can give those bills political momentum. It cannot enact them.
The cumulative picture is that the Executive Order is a strong opening move in a long process. The specific moves that will determine its impact are mostly downstream: agency action, sustained appropriations, supply chain build-out, clinical evidence generation, coverage decisions, and Congressional follow-through. Each of those is contingent. Each requires institutions and individuals that the Order can name but cannot direct.
That gap is the work of the next phase. It is also the work that the Ibogaine Healthcare Policy Institute exists to do. The next section discusses why.
Section 8: Where The Ibogaine Healthcare Policy Institute (IHPI) Fits
The work of translating the Executive Order into actual access for actual patients sits in a specific space. It is not the work of clinical research, which is being done by Compass, Definium, GH Research, AtaiBeckley/DemeRx, Gilgamesh, and the academic centers running their trials. It is not the work of state legislation, which Texas, Mississippi, Kentucky, Utah, Arizona, and others are advancing through their own political processes. It is not the work of federal regulation, which the FDA, DEA, HHS, and VA are now being asked to accelerate.
It is the work that sits between all of those, where coverage decisions, reimbursement codes, clinical guidelines, delivery blueprints, safety standards, and quality assurance frameworks have to be built so that an FDA-approved psychedelic medicine, when it arrives, actually reaches the patients it is designed to serve. The Executive Order does not address that space. The Ibogaine Healthcare Policy Institute (IHPI) was established to address these gaps for ibogaine.
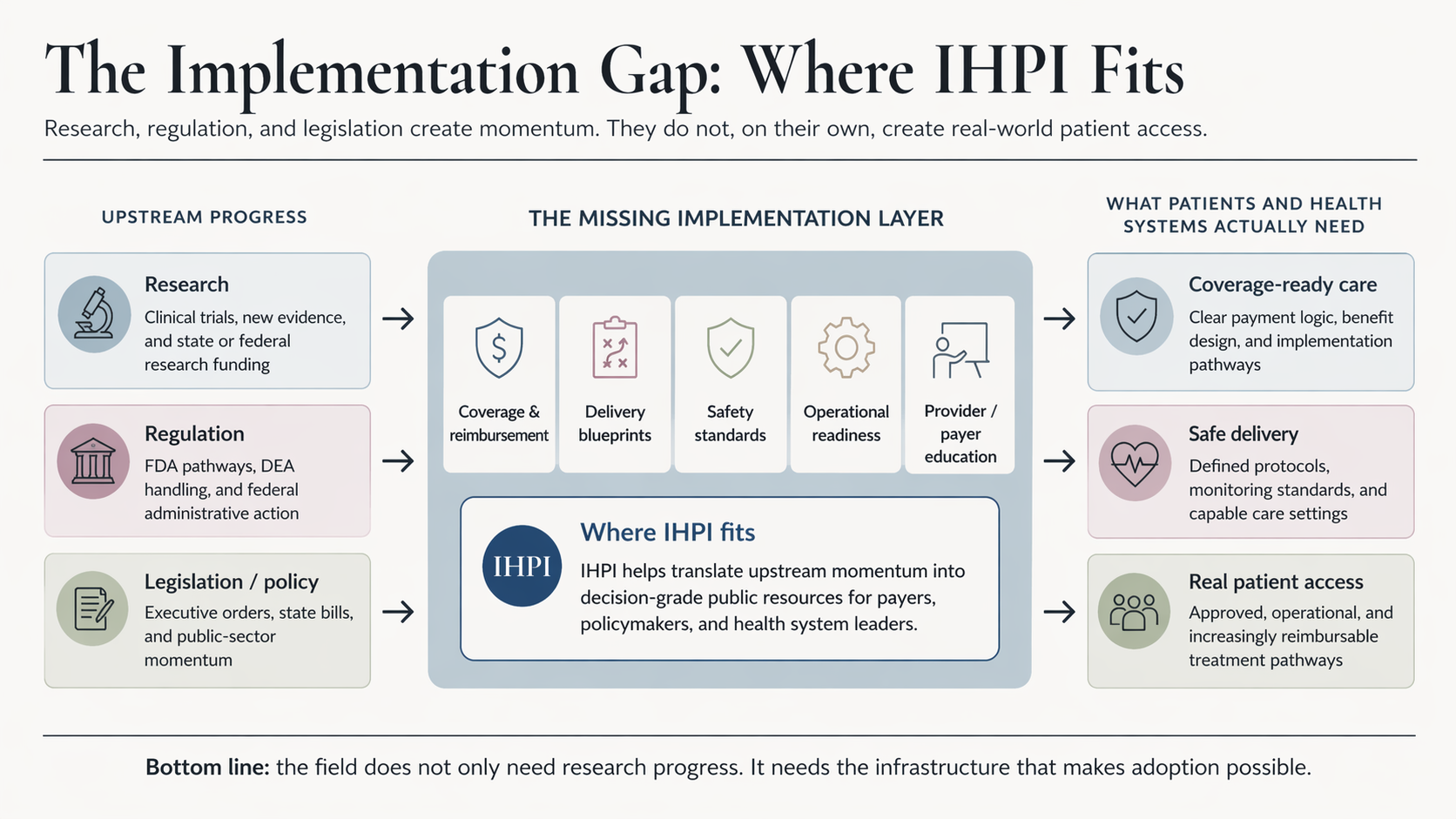
8.1 What IHPI is
IHPI is a neutral, evidence-based, implementation-focused nonprofit institute. Its mission is to move ibogaine from promise to policy to practice in healthcare. Its specific focus is the policy and payment infrastructure that will help ensure that evidence about ibogaine, as it accumulates, translates into responsible patient access.
The framing is deliberate. The institute does not advocate for ibogaine. It works on the policy, payment, and delivery questions that arise once clinical evidence is in hand and access decisions need to be made. That posture matters in a field where partisan and ideological framings have often dominated the public conversation. Implementation policy benefits from being treated as a technical challenge rather than a political one.
8.2 What IHPI does
IHPI’s work is organized into four interlocking areas, each corresponding directly to one of the unresolved questions identified earlier in this piece.
Coverage playbooks. The most concrete near-term gap is the absence of payer-ready coverage criteria for psychedelic-assisted therapy. IHPI develops the patient eligibility criteria, prior authorization rules, and medical coding pathways that payers and Medicaid programs need to make early coverage decisions. These are the inputs without which an FDA-approved psychedelic medicine cannot move from the prescription pad to the patient.
Health economics. Coverage decisions ultimately rest on actuarial models. IHPI builds the cost-offset analyses, utilization scenarios, and risk frameworks that payers can integrate into their own benefits, budgeting, and procurement workflows. For ibogaine specifically, the relevant economic comparison is not psychedelic-versus-psychedelic but rather the cost trajectory of standard treatment for opioid use disorder, treatment-resistant PTSD, and TBI, including the costs of repeated relapse, emergency care, and lost productivity that an effective intervention could potentially offset.
Delivery blueprints. Psychedelic-assisted therapy will not be delivered through standard primary care, at least in its early implementation. IHPI maps the residential, outpatient, and hospital pathways needed to deliver care safely, with the staffing models, screening protocols, monitoring requirements, and quality assurance processes that meet the demands of responsible delivery. For ibogaine, the cardiac safety profile makes delivery infrastructure particularly consequential. The space between a ketamine-clinic-like model and a more integrated medical model, which Section 6 discussed in the context of REMS, is precisely where delivery blueprint work has to happen.
Veterans-first pilots. The veteran population is, as Sections 4 and 5 discussed, both the most acute clinical need and the most politically aligned constituency for an early access program. IHPI’s pilot work is structured around the veterans cohort, in coordination with VA-adjacent partners and community providers. Pilots produce the operational evidence base that more general implementation requires, and they do so in a constituency where the case for responsible early access is strongest.
8.3 Why now
The Executive Order changes the timing of implementation work. The questions IHPI exists to answer were always going to need answering. They now need to be answered on a faster clock.
The federal posture established by the Order, the state research consortia coming online in Texas and elsewhere, the maturing pipeline of Compass, Usona, Transcend/Otsuka, Definium, GH Research, Helus/Cybin, and the ibogaine/noribogaine candidates, and the IND clearance for ibogaine announced on the day of the Order’s signing, together describe an environment where the gap between FDA approval and actual access could close faster than the implementation infrastructure currently supports. The cost of being unprepared for that timeline is concrete: patients who could have received care receive none, in the months and years between approval and the build-out of the systems that deliver care at scale. The first voucher awards sharpen this point: federal acceleration is already being applied selectively across the late-stage psychedelic and psychedelic-adjacent pipeline, while ibogaine-related development remains earlier and more dependent on Phase 1/2 safety generation.
The alternative is to do the work now. Coverage criteria, economic models, and delivery blueprints can be developed in parallel with clinical evidence generation. Payer engagement and provider network development can begin before approval rather than after. Safety standards and quality assurance frameworks can be defined in advance rather than improvised. None of this requires the federal government to move on a particular timeline. All of it benefits from the federal government having signaled, through the Executive Order, the direction it intends to move.
8.4 Who is involved
IHPI’s board brings the cross-sector experience that implementation work requires.
Andrea G. Barthwell, MD, DFASAM, is a past president and Distinguished Fellow of the American Society of Addiction Medicine and former Deputy Director for Demand Reduction at the White House Office of National Drug Control Policy. Her career combines clinical leadership, federal drug policy, and program implementation across treatment settings.
Andrea G. Barthwell, MD, DFASAM, is a past president and Distinguished Fellow of the American Society of Addiction Medicine and former Deputy Director for Demand Reduction at the White House Office of National Drug Control Policy. Her career combines clinical leadership, federal drug policy, and program implementation across treatment settings.
Leith States, MD, MPH, MBA, FACPM, is a board-certified preventive medicine physician who has served as Acting Assistant Secretary for Health and Chief Medical Officer in the Office of the Assistant Secretary for Health at HHS. His federal experience spans behavioral health, substance use policy, and cross-agency public health strategy. Prior to joining HHS, he spent nine years on active duty as a Navy Medical Officer serving in roles including Battalion Surgeon of a Marine infantry battalion, Public Health Emergency Officer for Navy Medicine West, and Officer in Charge of a Forward Deployable Preventive Medicine Unit
Eric Letsinger is the founder of Quantified Ventures and a strategic advisor to the outcomes-based firm that helps clients develop and finance initiatives that deliver measurable health, social, and environmental impact. He served as QV’s CEO from 2014 to 2023. Eric is a “tri-sector” executive, bringing 25+ years of leadership experience in government, nonprofit, and private-sector organizations operating in healthcare, the environment, education, and housing. He has led transformative, public-private initiatives to drive social impact in complex, cross-sector business environments, including IBM, Baltimore Public Schools, Baltimore Housing Department, Cyveillance Software, PWC, and Samaritan Inn’s Homeless Services.
The combination is intentional. Implementation policy in this space requires fluency in clinical addiction medicine, payer and benefit design, federal health agency operations, psychedelic-specific stewardship, and experience developing large-scale public-private partnerships. Each of those competencies is represented.
In addition to the Board, IHPI has a deep bench of staff and advisors who bring a wealth of deep and relevant expertise to the institute’s work. This includes, among others:
Eric Bailly, MA, LPC, LADC, founded NorthStar Behavioral Health Advisory and previously led the Alliance for Addiction Payment Reform at Third Horizon Strategies. He spent fifteen years at Elevance Health (Anthem) advancing enterprise substance use disorder and opioid strategy. His expertise sits squarely in the payer policy and benefit design questions that IHPI’s coverage playbook depends on.
Kenneth Tupper, PhD, is a Collaborating Scientist with the Canadian Institute for Substance Use Research and an Adjunct Professor at the University of Victoria, with prior leadership roles at the British Columbia Ministry of Health and the BC Centre on Substance Use. His work focuses on evidence-informed psychedelic policy and stewardship for innovation across healthcare systems.
Ryan Roberts, MBA, is a USMC combat veteran and healthcare executive with two decades of leadership across community, hospital, and academic medicine. As the founder of The Journey Home, Ryan develops peer-driven restoration frameworks for moral injury, bridging the gap between lived experience and institutional strategy. He serves as a clinical research partner with Baylor College of Medicine and the Menninger Clinic on veteran psychedelic therapy studies and advocates for legislative change as a founding member of the Veterans Mental Health Leadership Coalition. Ryan is dedicated to ensuring veterans are not just recipients of care, but active architects of the policies and programs that serve them.
Lia Mix, LMFT, CPTR, is IHPI’s acting Executive Director as well as the Founder and CEO of Delphi, a consulting firm dedicated to the healthy growth of the psychedelic movement. With over 20 years of experience as a licensed marriage and family therapist (LMFT) and healthcare strategist, she specializes in integrating novel therapies into insurance systems. Lia is certified in Psychedelic Therapies and Research (CPTR) by the California Institute for Integral Studies and has been instrumental in establishing the core infrastructure for integrating psychedelic-assisted therapy into the US healthcare system.
Floris Wolswijk is a healthcare innovator with a background in psychology and a deep understanding of the psychedelic ecosystem. He has been pivotal in advancing healthcare solutions in the field of psychedelics. In addition to his consulting work at Delphi, Floris is the founder of Blossom, the most comprehensive database for psychedelic medicine insights.
David Abernathy is an executive and nonprofit leader with more than a decade and a half of experience in drug policy reform and more than two decades in nonprofit governance. He serves as Chief Strategy Officer at Delphi and serves on the board of numerous nonprofit and for-profit companies, including The Marijuana Policy Project and The Arcview Group. In 2013, he launched and led Arcview Market Research, a cannabis-focused market research firm that has published thousands of pages of reports covering every legal, medical, and adult-use cannabis market worldwide.
8.5 Engagement
IHPI is actively engaging with payers and Medicaid programs, health systems and provider networks, veterans organizations and federal partners, philanthropic and policy funders, and therapeutic developers and service providers. The work ahead includes the publication of the initial coverage playbook, development of a health economics model, the first round of delivery blueprint pilots, and active stakeholder engagement across the policy and payment ecosystem.
For organizations whose work intersects with the implementation gap this Executive Order has highlighted, IHPI is open to partnership, advisory engagement, and collaborative development of the policy and payment infrastructure required for responsible psychedelic medicine adoption.
Section 9: Sources and Further Reading
The piece above draws on a wide range of policy documents, scientific publications, regulatory filings, news coverage, and expert analysis. The list below is curated rather than comprehensive. It groups the most useful sources by topic and provides a brief orientation to what each grouping offers, for readers who want to go deeper on any particular thread.
The Executive Order itself and the early reactions to it
Reading the Order in full is short and worth it. The accompanying White House Fact Sheet provides the administration’s framing of the policy goals. For substantive legal and policy commentary, the Petrie-Flom Center Q&A with I. Glenn Cohen and Mason Marks, published the day of the signing, is an analytically rigorous piece. Psychedelic Alpha’s analysis is a useful industry read, with particular attention to implementation questions.
- Executive Order, Accelerating Medical Treatments for Serious Mental Illness, April 18, 2026: https://www.whitehouse.gov/presidential-actions/2026/04/accelerating-medical-treatments-for-serious-mental-illness/
- White House Fact Sheet on the Executive Order: https://www.whitehouse.gov/fact-sheets/2026/04/fact-sheet-president-donald-j-trump-is-accelerating-medical-treatments-for-serious-mental-illness/
- FDA, ‘FDA Accelerates Action on Treatments for Serious Mental Illness Following Executive Order,’ April 24, 2026: https://www.fda.gov/news-events/press-announcements/fda-accelerates-action-treatments-serious-mental-illness-following-executive-order
- Petrie-Flom Center, “A New Executive Order on Psychedelics: Q&A with I. Glenn Cohen and Mason Marks”: https://petrieflom.law.harvard.edu/2026/04/18/a-new-executive-order-on-psychedelics-q-a-with-i-glenn-cohen-and-mason-marks/
- Psychedelic Alpha, “President Trump Signs Executive Order to Accelerate Psychedelic Research and Access”: https://psychedelicalpha.com/news/president-trump-signs-executive-order-to-accelerate-psychedelic-research-and-access/
- CNBC, “How Trump’s psychedelics executive order could unlock stalled cannabis reform”: https://www.cnbc.com/2026/04/20/trump-psychedelics-executive-order-cannabis-reform.html
- CNN, “Trump signs executive order urging more research into ibogaine and other psychedelics”: https://www.cnn.com/2026/04/18/politics/trump-signs-executive-order-urging-more-research-into-psychedelic-ibogaine
- PBS NewsHour, “Trump signs order to hasten review of psychedelics”: https://www.pbs.org/newshour/politics/trump-signs-order-to-speed-review-of-psychedelics
The clinical and scientific evidence base
The Stanford magnesium-ibogaine work is the most clinically and methodologically substantial body of US-affiliated research on ibogaine. The 2024 Nature Medicine paper documents the primary observational study; the 2025 Nature Mental Health follow-up reports neuroimaging findings. The earlier observational work by The Mission Within and collaborators, published in Chronic Stress in 2020, extends the evidence base for psychedelic-assisted treatment in Special Operations Forces veterans. For ibogaine in opioid use disorder specifically, the systematic review in Current Neuropharmacology (2023) is the cleanest single entry point.
- Cherian et al., “Magnesium–ibogaine therapy in veterans with traumatic brain injuries,” Nature Medicine (January 2024): https://www.nature.com/articles/s41591-023-02705-w
- Stanford Medicine summary of the 2024 study and 2025 follow-up: https://med.stanford.edu/news/all-news/2024/01/ibogaine-ptsd.html
- US Medicine on the Nature Mental Health follow-up study (July 2025): https://www.usmedicine.com/clinical-topics/tbi/magnesium-ibogaine-therapy-could-offer-treatment-option-for-tbi/
- Davis et al., “Psychedelic Treatment for Trauma-Related Psychological and Cognitive Impairment Among US Special Operations Forces Veterans,” Chronic Stress (2020): https://journals.sagepub.com/doi/full/10.1177/2470547020939564
- Mosca et al., “Ibogaine/noribogaine in the treatment of substance use disorders: A systematic review,” Current Neuropharmacology (2023): https://pubmed.ncbi.nlm.nih.gov/37936434/
- Noller, Frampton & Yazar-Klosinski, “Ibogaine treatment outcomes for opioid dependence from a twelve-month follow-up observational study,” American Journal of Drug and Alcohol Abuse (2018): https://pubmed.ncbi.nlm.nih.gov/29225508/
- Veterans Exploring Treatment Solutions (VETS), Stanford Brain Stimulation Lab Ibogaine Study page: https://vetsolutions.org/research/stanford-university-neuroimaging-study/
- The Mission Within, News & Research: https://missionwithin.org/news-research/
Federal access pathways and regulatory context
The Right to Try Act and the FDA’s expanded access mechanisms are the two patient-facing federal access pathways. The FDA’s plain-English pages on each are the most reliable starting points. For the legal context the EO seeks to address, the Ninth Circuit’s 2025 ruling against Dr. Sunil Aggarwal is the relevant case. The cannabis Executive Order from December 2025 is the most relevant comparison for thinking about implementation timelines, and the Biden October 2022 marijuana directive provides a longer-horizon precedent for how presidential drug-policy directives actually move through agencies.
- FDA, “Right to Try” patient information page: https://www.fda.gov/patients/learn-about-expanded-access-and-other-treatment-options/right-try
- FDA, “Expanded Access” public health focus page: https://www.fda.gov/news-events/public-health-focus/expanded-access
- Drug & Device Law Blog on the Ninth Circuit Right to Try ruling (February 2025): https://www.druganddevicelawblog.com/2025/02/ninth-circuit-rejects-right-to-try-hallucinogenic-drugs.html
- Executive Order 14370, Encouraging Additional Research on Marijuana (December 18, 2025): https://www.whitehouse.gov/presidential-actions/2025/12/encouraging-additional-research-on-marijuana/
- White House article on the HALT Fentanyl Act (July 2025): https://www.whitehouse.gov/articles/2025/07/halt-fentanyl-act/
- White House statement on Biden marijuana reform directive (October 2022): https://www.whitehouse.gov/briefing-room/statements-releases/2022/10/06/statement-from-president-biden-on-marijuana-reform/
- Senator Booker press release on the Freedom to Heal Act: https://www.booker.senate.gov/news/press/booker-and-paul-introduce-bipartisan-bill-to-expand-access-to-experimental-treatments-for-terminally-ill-patients
- Military.com coverage of the Veterans Health Administration Novel Therapeutics Preparedness Act (S.4220): https://www.military.com/daily-news/2026/04/06/major-step-new-law-would-prep-va-fda-approved-psychedelic-treatments.html
State-level activity and the legislative landscape
For the Bryan Hubbard story, the Reason profile from September 2024 is the definitive long-form account. Rolling Stone’s April 2026 feature by Cassady Rosenblum extends the narrative through the Texas pivot, the Aspen Americans for Ibogaine conference, and the Gabonese delegation’s push for US Nagoya Protocol accession. Americans for Ibogaine, the advocacy nonprofit Hubbard now leads, hosts the OSINT report on the December 2023 collapse of his Kentucky initiative. For the broader state-by-state legislative picture, the Berkeley Center for the Science of Psychedelics Law and Policy Map is one of the most comprehensive single resources. Psychedelic Alpha’s 2025 state policy surge analysis is the most useful narrative overview.
- Reason, “The Psychedelic Emancipator of Kentucky”: https://reason.com/2024/09/15/the-psychedelic-emancipator-of-kentucky/
- Americans for Ibogaine: https://www.americansforibogaine.org/
- Reveille Advisors / Americans for Ibogaine OSINT report on the Kentucky 2023 collapse: https://www.americansforibogaine.org/news/reveille-advisors-kentucky/
- Berkeley Center for the Science of Psychedelics, Law and Policy Map: https://psychedelics.berkeley.edu/law-and-policy-map
- Psychedelic Alpha, “2025’s Psychedelic Policy Surge: A State-by-State, Bill-by-Bill Analysis”: https://psychedelicalpha.com/news/2025s-psychedelic-policy-surge-a-state-by-state-bill-by-bill-analysis/
- High Times, “Psychedelic Reform Is Spreading Faster Than Anyone Expected”: https://hightimes.com/psychedelics/psychedelic-reform-is-spreading-faster-than-anyone-expected-the-movement-is-trying-not-to-blow-it/
- Rolling Stone, Cassady Rosenblum, “How Ibogaine Became the Darling of the Psychedelic Right” (April 22, 2026): https://www.rollingstone.com/culture/culture-features/ibogaine-psychedelic-right-ptsd-addiction-iboga-1235544846/
Supply chain, sourcing, and the international context
The Gabon iboga supply chain is the most underdiscussed dependency in the entire ibogaine policy conversation, and the picture is wider than just iboga. Filament Health’s May 2023 announcement of the first Nagoya-compliant export is a milestone document. Dr. Joseph Barsuglia’s August 2025 sourcing presentation to the Colorado Natural Medicine Board is the clearest single explanation of how ibogaine is actually produced today, including the Voacanga africana pathway that supplies most current clinical material. The Lucid News piece on the Gabon-side process and the Georgetown Journal of International Law analysis on the Nagoya Protocol’s role in psychedelic supply ethics provide the deeper context for understanding how international law shapes what is possible.
- Filament Health, “First Ever Nagoya Protocol-Compliant Shipment of Iboga from Gabon” (May 2023): https://www.filament.health/news/filament-health-announces-first-ever-nagoya-protocol-compliant-shipment-of-iboga-from-gabon
- Lucid News, “Gabon Takes First Step Toward Legal Export of Sustainable Iboga”: https://www.lucid.news/gabon-takes-first-step-toward-legal-export-of-sustainable-iboga/
- Georgetown Journal of International Law, “The Promise of Nagoya: Indigenous Reciprocity in the Psychedelic Renaissance”: https://www.law.georgetown.edu/international-law-journal/blog/the-promise-of-nagoya-indigenous-reciprocity-in-the-psychedelic-renaissance/
- Dr. Joseph Barsuglia, “Iboga and Ibogaine Sourcing” presentation to the Colorado Natural Medicine Board (August 2025): https://www.josephbarsuglia.com/s/iboga-and-ibogaine-Sourcing-production.pdf
Veterans, opioid use disorder, and the underlying need
The CDC and KFF data resources are the most reliable starting points for understanding the opioid crisis trajectory and the 2024 decline. The first-term Trump Executive Order 13822, issued in January 2018, provides direct continuity with the current administration’s posture on veteran mental health. For organizations doing direct work with veterans accessing psychedelic-assisted treatment, VETS, The Mission Within, and Heroic Hearts Project are the three most established.
- CDC press release on 27% decline in 2024 overdose deaths: https://www.cdc.gov/media/releases/2025/2025-cdc-reports-decline-in-us-drug-overdose-deaths.html
- KFF analysis of national opioid overdose patterns: https://www.kff.org/mental-health/opioid-overdose-deaths-national-trends-and-variation-by-demographics-and-states/
- CDC Overdose Prevention data resources (April 2026 update): https://www.cdc.gov/overdose-prevention/data-research/facts-stats/index.html
- Executive Order 13822, Supporting Our Veterans During Their Transition from Uniformed Service to Civilian Life (January 2018): https://trumpwhitehouse.archives.gov/presidential-actions/presidential-executive-order-supporting-veterans-transition-uniformed-service-civilian-life/
Where the institute fits
For readers interested in the Ibogaine Healthcare Policy Institute and its work, the institute’s page at Delphi is the place to start. The wider Delphi site provides context on the firm’s work in psychedelic policy, education, and consulting more broadly.
- Ibogaine Healthcare Policy Institute (IHPI): https://delphi-circle.com/ihpi/
- Delphi: https://delphi-circle.com/
- Contact and partnership inquiries: https://delphi-circle.com/contact/
